Merge samples
Mechthild Lütge
14 May 2020
Last updated: 2022-09-12
Checks: 6 1
Knit directory: humanCardiacFibroblasts/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20210903) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 9b017be. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/GSEA/
Ignored: data/humanFibroblast/
Ignored: figure/DEgenesGZplusSG_Groups.Rmd/.DS_Store
Untracked files:
Untracked: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 20-1.pdf
Untracked: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 20-1.png
Unstaged changes:
Modified: analysis/integrateAcrossPatientsGZplusSG.Rmd
Modified: analysis/mergeHumanSamplesPlusGraz.Rmd
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 0-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 0-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 1-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 1-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 10-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 10-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 11-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 11-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 12-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 12-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 13-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 13-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 14-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 14-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 15-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 15-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 16-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 16-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 17-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 17-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 18-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 18-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 19-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 19-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 2-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 2-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 3-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 3-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 4-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 4-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 5-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 5-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 6-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 6-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 7-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 7-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 8-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 8-1.png
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 9-1.pdf
Modified: figure/mergeHumanSamplesPlusGraz.Rmd/top marker 9-1.png
Modified: metadata2.txt
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown
(analysis/integrateAcrossPatientsGZplusSG.Rmd) and HTML
(docs/integrateAcrossPatientsGZplusSG.html) files. If
you’ve configured a remote Git repository (see
?wflow_git_remote), click on the hyperlinks in the table
below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 02765dc | mluetge | 2022-07-19 | GSEA across diff groups |
| Rmd | 3e98bf3 | mluetge | 2022-07-15 | run DE genes HH vs Myo |
| html | 3e98bf3 | mluetge | 2022-07-15 | run DE genes HH vs Myo |
| Rmd | d9e1a98 | mluetge | 2022-07-04 | integrate samples Graz |
| html | d9e1a98 | mluetge | 2022-07-04 | integrate samples Graz |
load packages
suppressPackageStartupMessages({
library(SingleCellExperiment)
library(tidyverse)
library(Seurat)
library(magrittr)
library(dplyr)
library(purrr)
library(ggplot2)
library(here)
library(runSeurat3)
library(ggsci)
library(ggpubr)
library(pheatmap)
library(viridis)
library(sctransform)
})integrate data
basedir <- here()
seurat <- readRDS(file = paste0(basedir,
"/data/humanHeartsPlusGraz_merged_seurat.rds"))
## integrate data across patients
Idents(seurat) <- seurat$ID
seurat.list <- SplitObject(object = seurat, split.by = "ID")
for (i in 1:length(x = seurat.list)) {
seurat.list[[i]] <- NormalizeData(object = seurat.list[[i]],
verbose = FALSE)
seurat.list[[i]] <- FindVariableFeatures(object = seurat.list[[i]],
selection.method = "vst", nfeatures = 500, verbose = FALSE)
}
seurat.anchors <- FindIntegrationAnchors(object.list = seurat.list, dims = 1:20)
seurat.int <- IntegrateData(anchorset = seurat.anchors, dims = 1:20)
DefaultAssay(object = seurat.int) <- "integrated"
# rerun seurat
seurat.int <- ScaleData(object = seurat.int, verbose = FALSE)
seurat.int <- RunPCA(object = seurat.int, npcs = 20, verbose = FALSE)
seurat.int <- RunTSNE(object = seurat.int, reduction = "pca", dims = 1:20)
seurat.int <- RunUMAP(object = seurat.int, reduction = "pca", dims = 1:20)
seurat.int <- FindNeighbors(object = seurat.int, reduction = "pca", dims = 1:20,
k.param = 50)
res <- c(0.6,0.8,0.4,0.25)
for(i in 1:length(res)){
seurat.int <- FindClusters(object = seurat.int, resolution = res[i],
random.seed = 1234)
}Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 52908
Number of edges: 6183319
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.9212
Number of communities: 17
Elapsed time: 25 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 52908
Number of edges: 6183319
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.9041
Number of communities: 18
Elapsed time: 27 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 52908
Number of edges: 6183319
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.9424
Number of communities: 15
Elapsed time: 28 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 52908
Number of edges: 6183319
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.9610
Number of communities: 12
Elapsed time: 25 secondsDefaultAssay(object = seurat.int) <- "RNA"
seurat <- seurat.int
remove(seurat.int)
seurat$seurat_clusters <- seurat$integrated_snn_res.0.25
Idents(seurat) <- seurat$seurat_clusterssave int object
saveRDS(seurat, file = paste0(basedir,
"/data/humanHeartsPlusGraz_intPatients_merged",
"_seurat.rds"))color vectors
colPal <- pal_igv()(length(levels(seurat)))
colTec <- pal_jama()(length(unique(seurat$technique)))
colSmp <- c(pal_uchicago()(8), pal_npg()(8), pal_aaas()(10))[1:length(unique(seurat$dataset))]
colCond <- pal_npg()(length(unique(seurat$cond)))
colID <- c(pal_jco()(10), pal_npg()(10))[1:length(unique(seurat$ID))]
colOrig <- pal_aaas()(length(unique(seurat$origin)))
colIso <- pal_nejm()(length(unique(seurat$isolation)))
colProc <- pal_aaas()(length(unique(seurat$processing)))
names(colPal) <- levels(seurat)
names(colTec) <- unique(seurat$technique)
names(colSmp) <- unique(seurat$dataset)
names(colCond) <- unique(seurat$cond)
names(colID) <- unique(seurat$ID)
names(colOrig) <- unique(seurat$origin)
names(colIso) <- unique(seurat$isolation)
names(colProc) <- unique(seurat$processing)vis data
clusters
DimPlot(seurat, reduction = "umap", cols=colPal)+
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank(),
panel.grid.minor = element_blank()) +
xlab("UMAP1") +
ylab("UMAP2")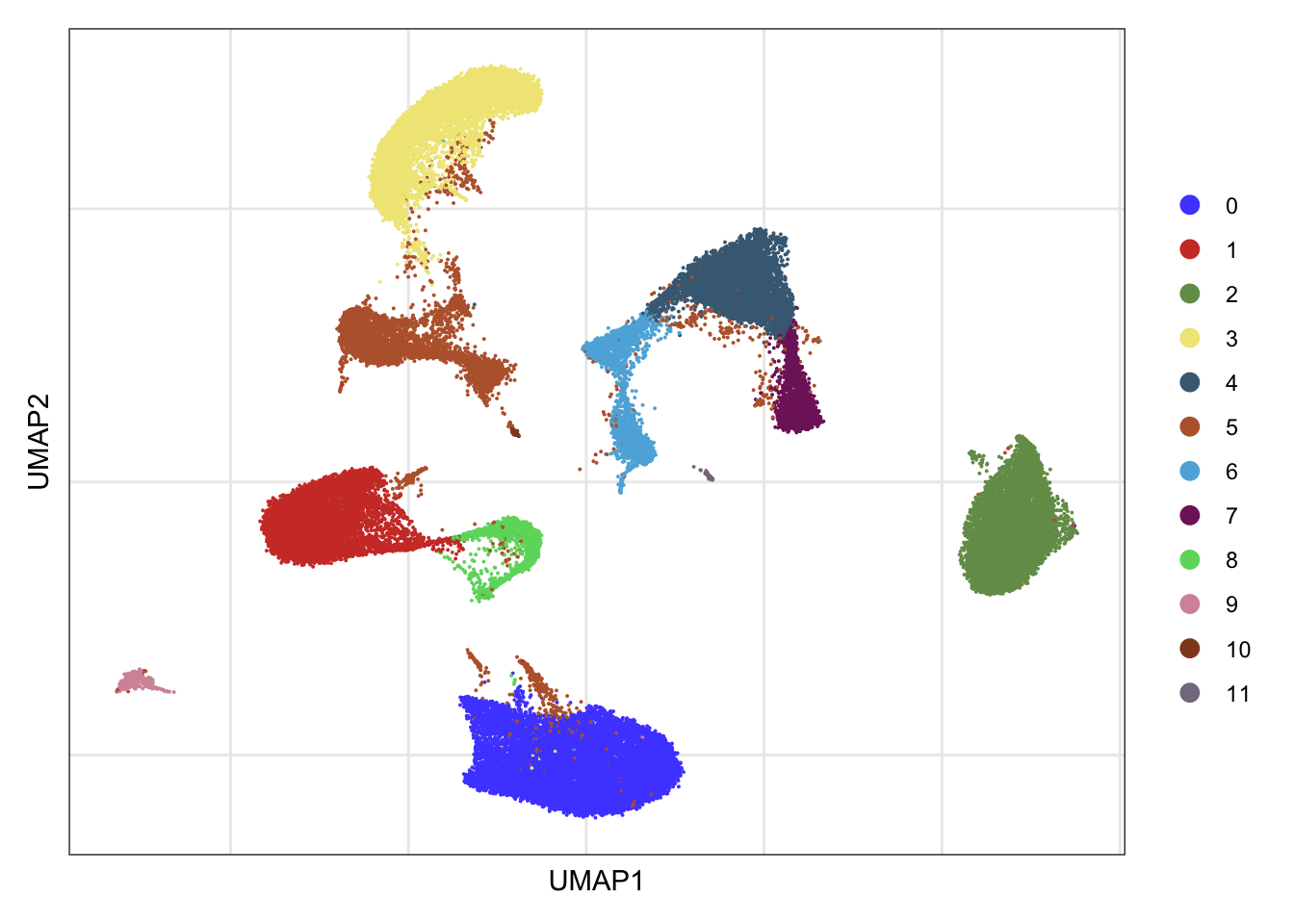
technique
DimPlot(seurat, reduction = "umap", group.by = "technique", cols=colTec)+
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank(),
panel.grid.minor = element_blank()) +
xlab("UMAP1") +
ylab("UMAP2")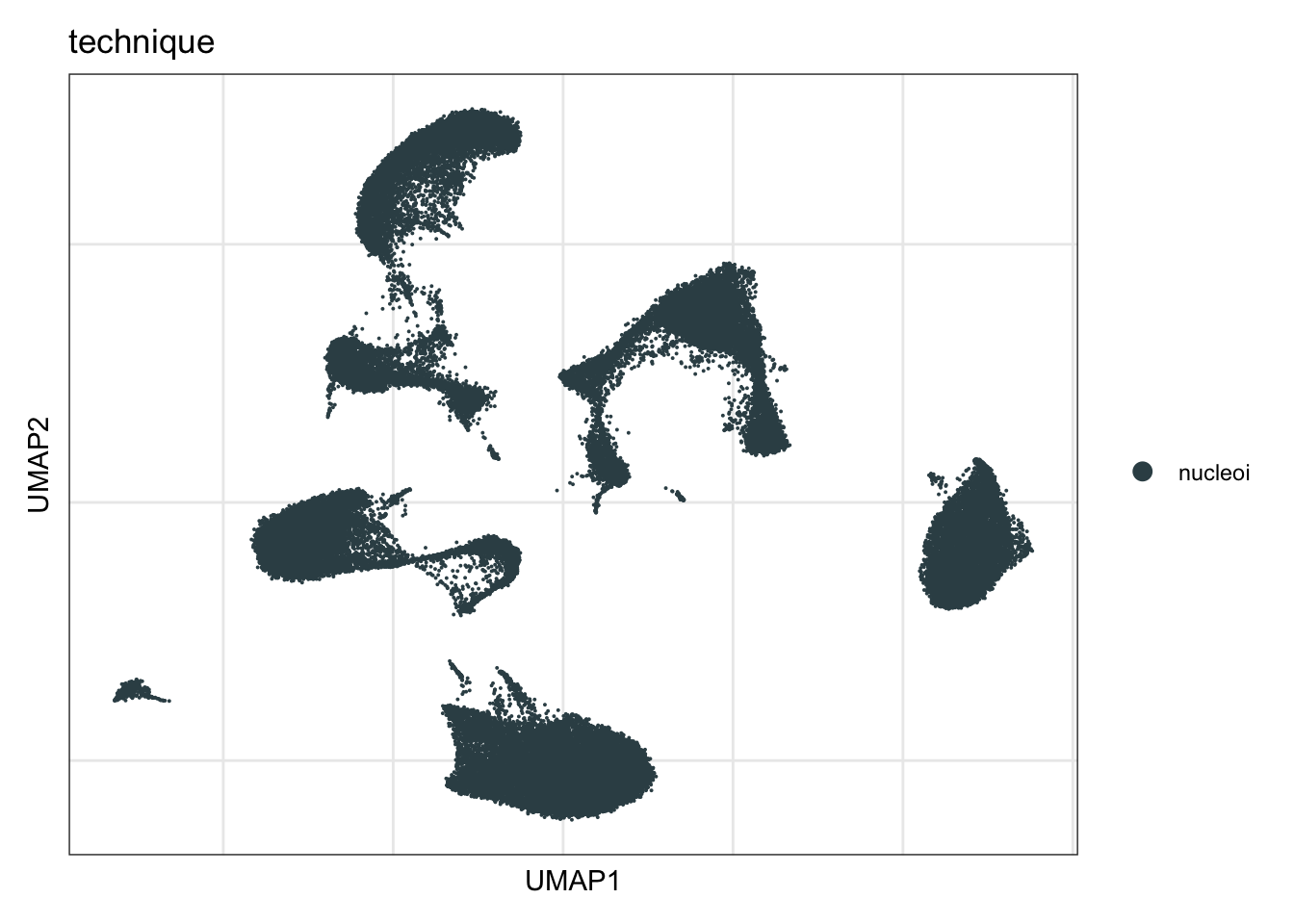
Sample
DimPlot(seurat, reduction = "umap", group.by = "dataset", cols=colSmp)+
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank(),
panel.grid.minor = element_blank()) +
xlab("UMAP1") +
ylab("UMAP2")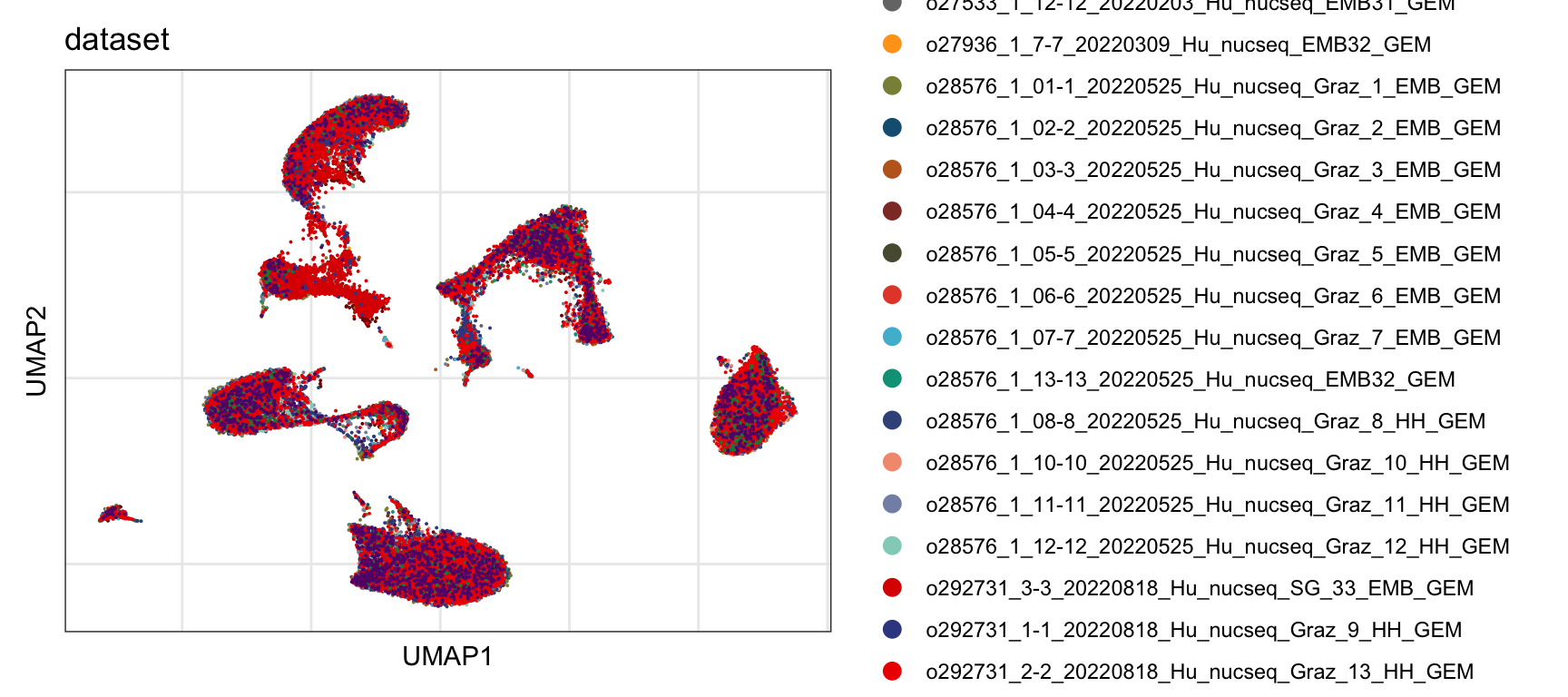
ID
DimPlot(seurat, reduction = "umap", group.by = "ID", cols=colID)+
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank(),
panel.grid.minor = element_blank()) +
xlab("UMAP1") +
ylab("UMAP2")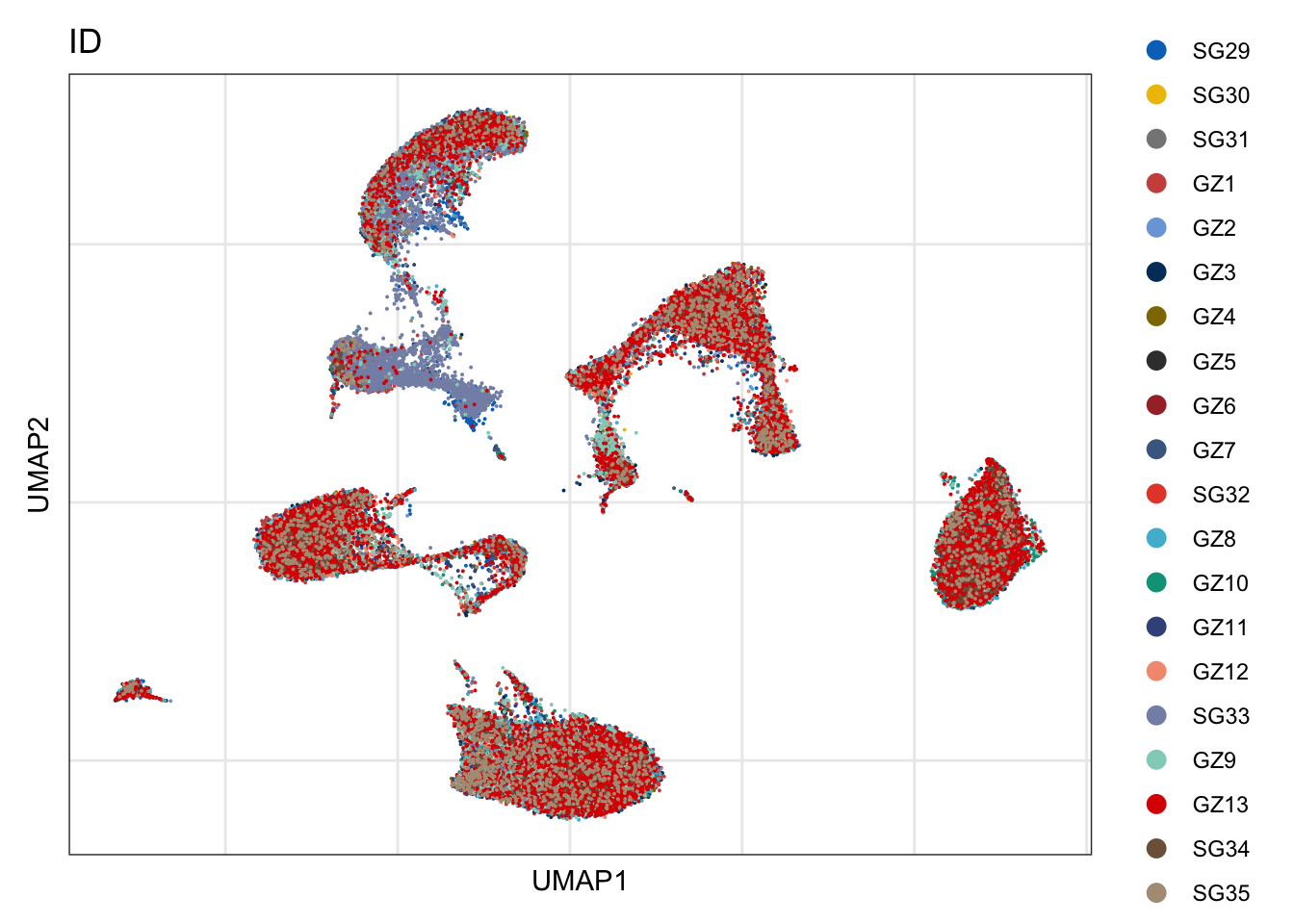
Origin
DimPlot(seurat, reduction = "umap", group.by = "origin", cols=colOrig)+
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank(),
panel.grid.minor = element_blank()) +
xlab("UMAP1") +
ylab("UMAP2")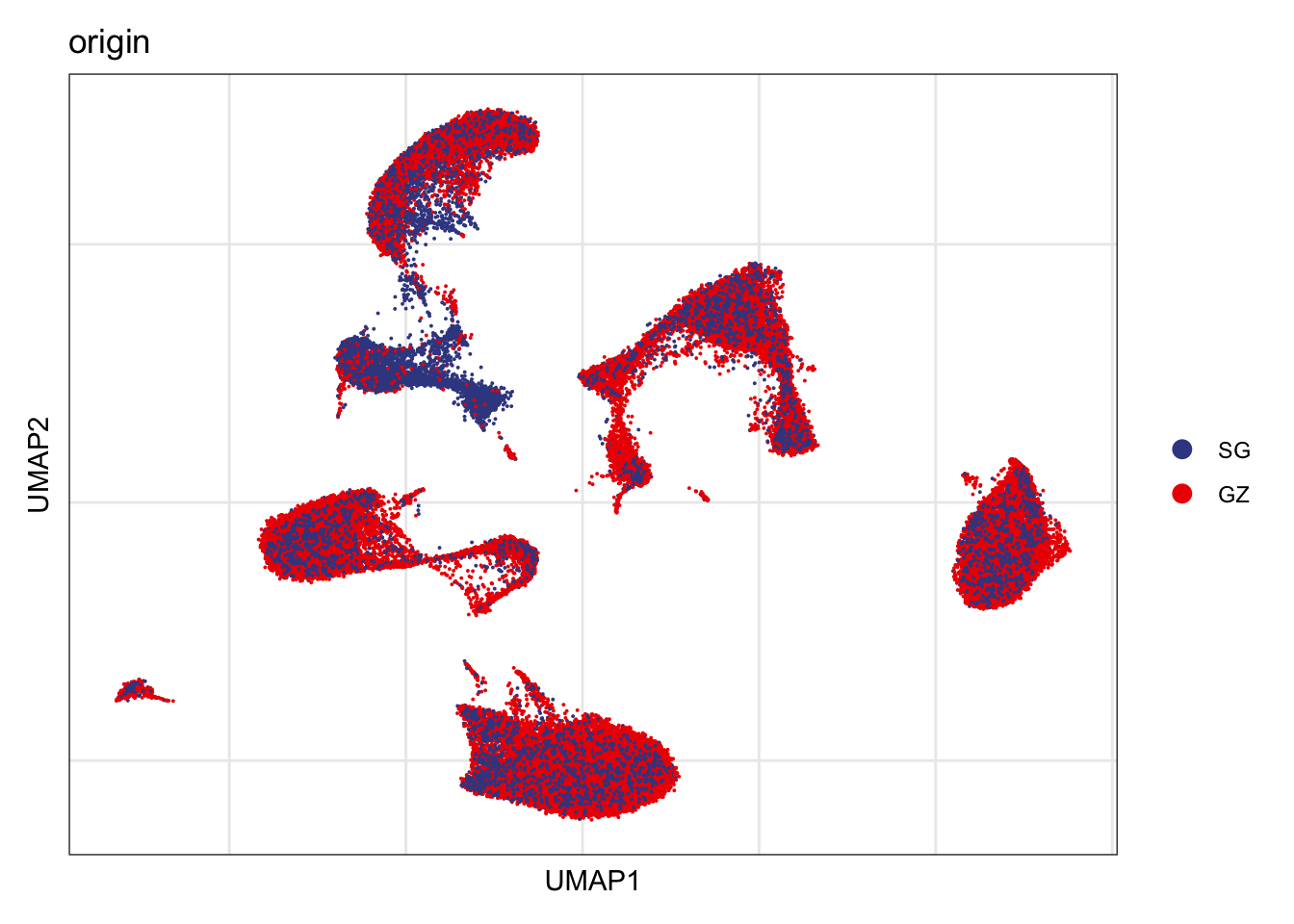
isolation
DimPlot(seurat, reduction = "umap", group.by = "isolation", cols=colIso)+
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank(),
panel.grid.minor = element_blank()) +
xlab("UMAP1") +
ylab("UMAP2")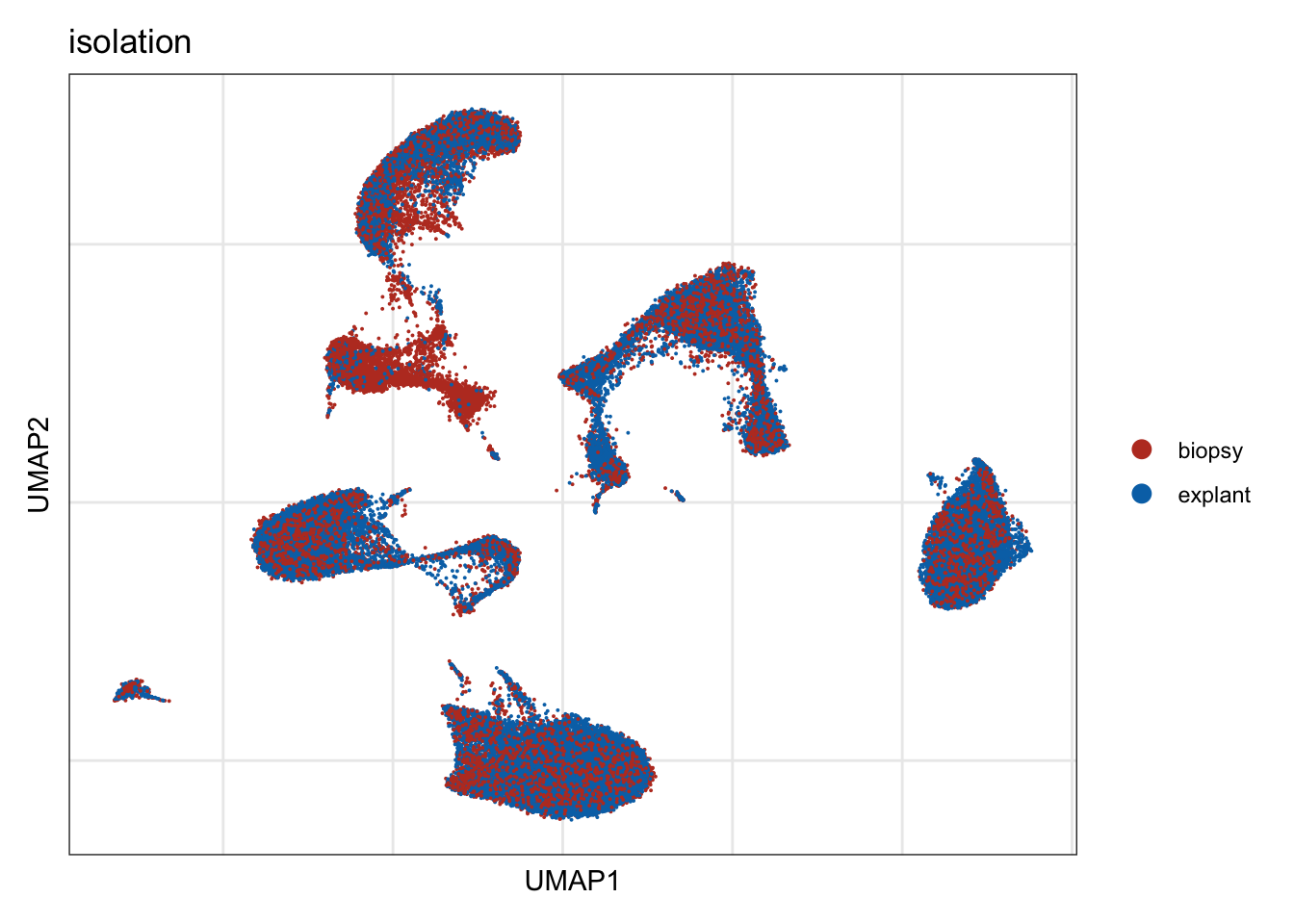
cond
DimPlot(seurat, reduction = "umap", group.by = "cond", cols=colCond)+
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank(),
panel.grid.minor = element_blank()) +
xlab("UMAP1") +
ylab("UMAP2")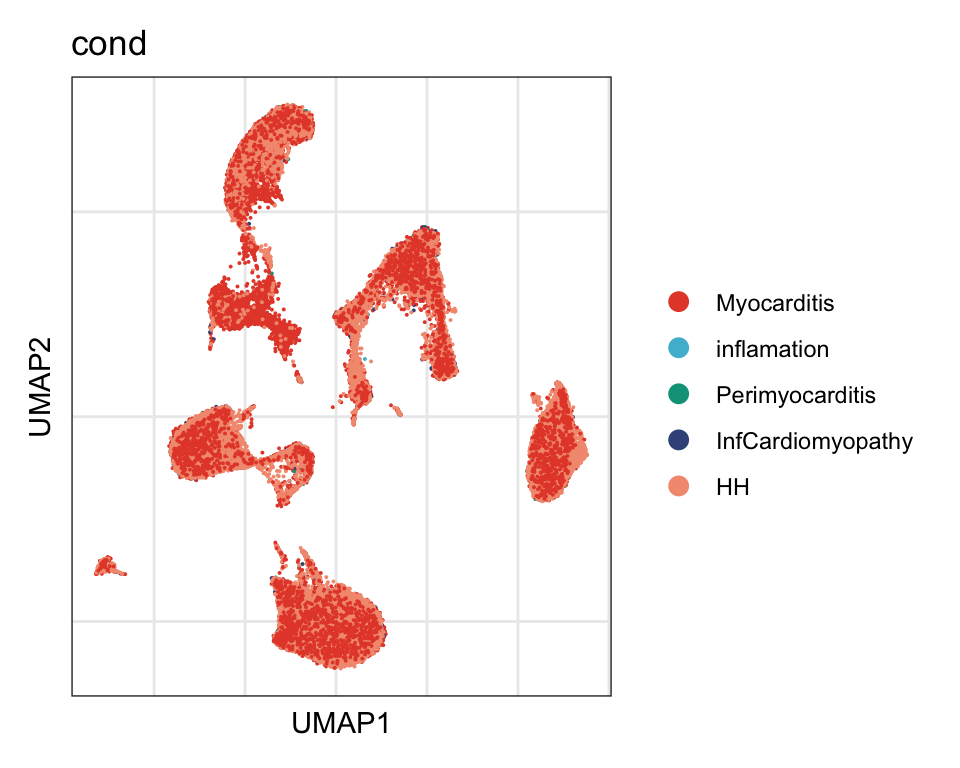
processing
DimPlot(seurat, reduction = "umap", group.by = "processing", cols=colProc)+
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank(),
panel.grid.minor = element_blank()) +
xlab("UMAP1") +
ylab("UMAP2")
marker genes
seurat_markers_all <- FindAllMarkers(object = seurat, assay ="RNA",
only.pos = TRUE, min.pct = 0.25,
logfc.threshold = 0.25,
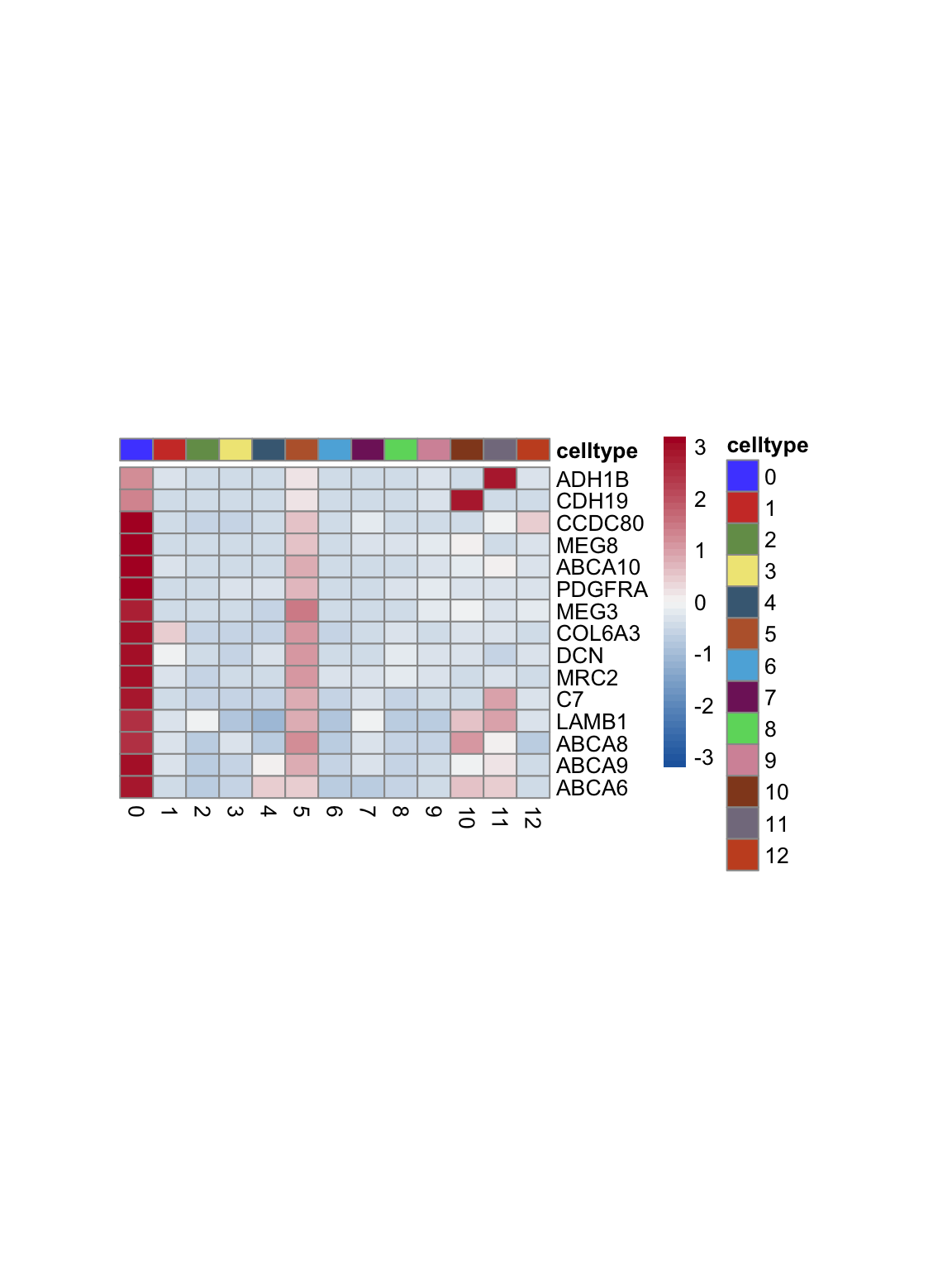
test.use = "wilcox")top 15 marker genes per cluster
cluster <- levels(seurat)
selGenesAll <- seurat_markers_all %>% group_by(cluster) %>%
top_n(-15, p_val_adj) %>%
top_n(15, avg_log2FC)
selGenesAll <- selGenesAll %>% mutate(geneIDval=gsub("^.*\\.", "", gene)) %>% filter(nchar(geneIDval)>1)
template_hm <- c(
"#### {{cl}}\n",
"```{r top marker {{cl}}, fig.height=8, fig.width=6, echo = FALSE}\n",
"selGenes <- selGenesAll %>% filter(cluster=='{{cl}}')",
"pOut <- avgHeatmap(seurat = seurat, selGenes = selGenes,
colVecIdent = colPal,
ordVec=levels(seurat),
gapVecR=NULL, gapVecC=NULL,cc=FALSE,
cr=T, condCol=F)\n",
"```\n",
"\n"
)
plots_gp <- lapply(cluster,
function(cl) knitr::knit_expand(text = template_hm)
)
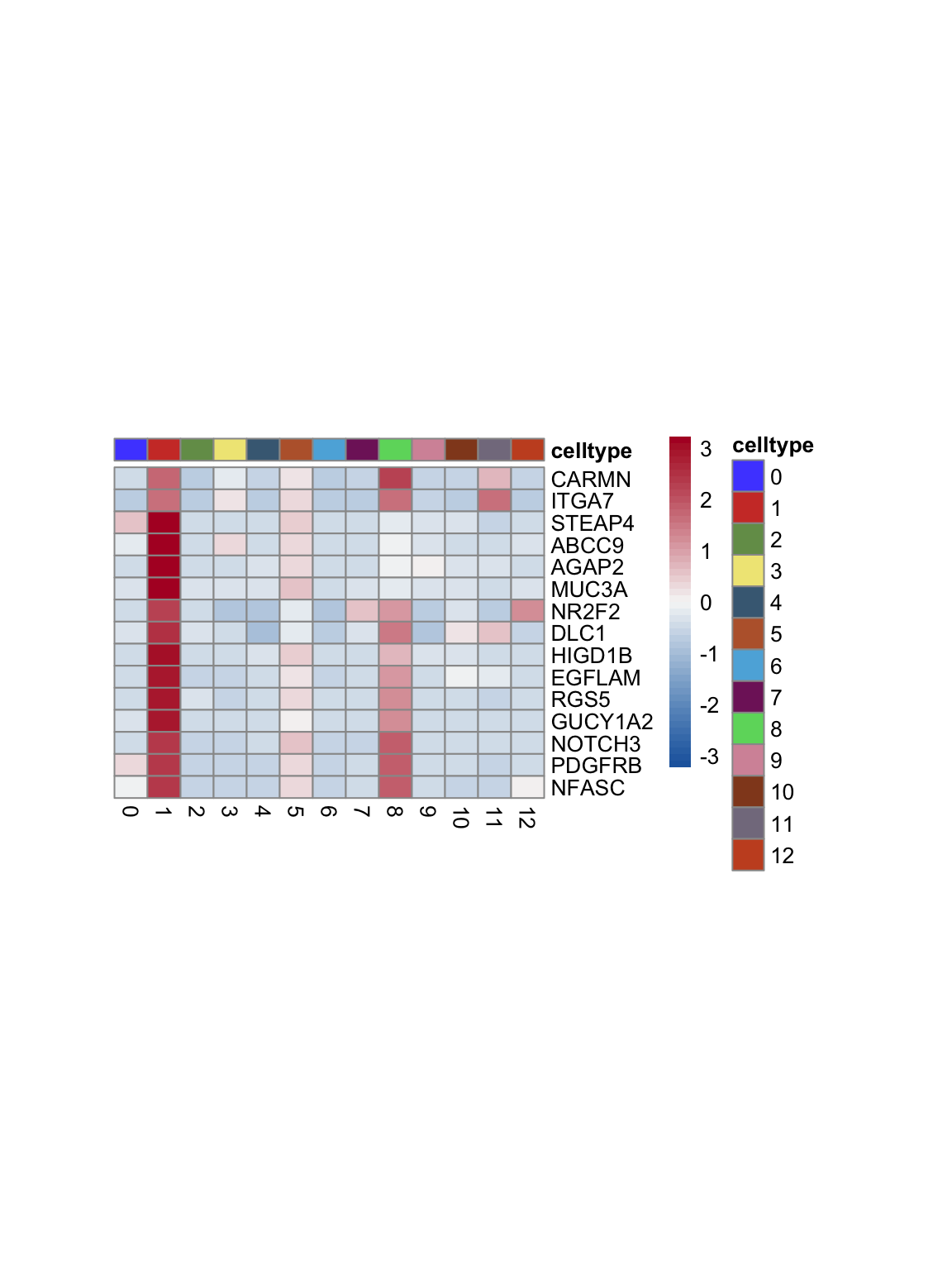
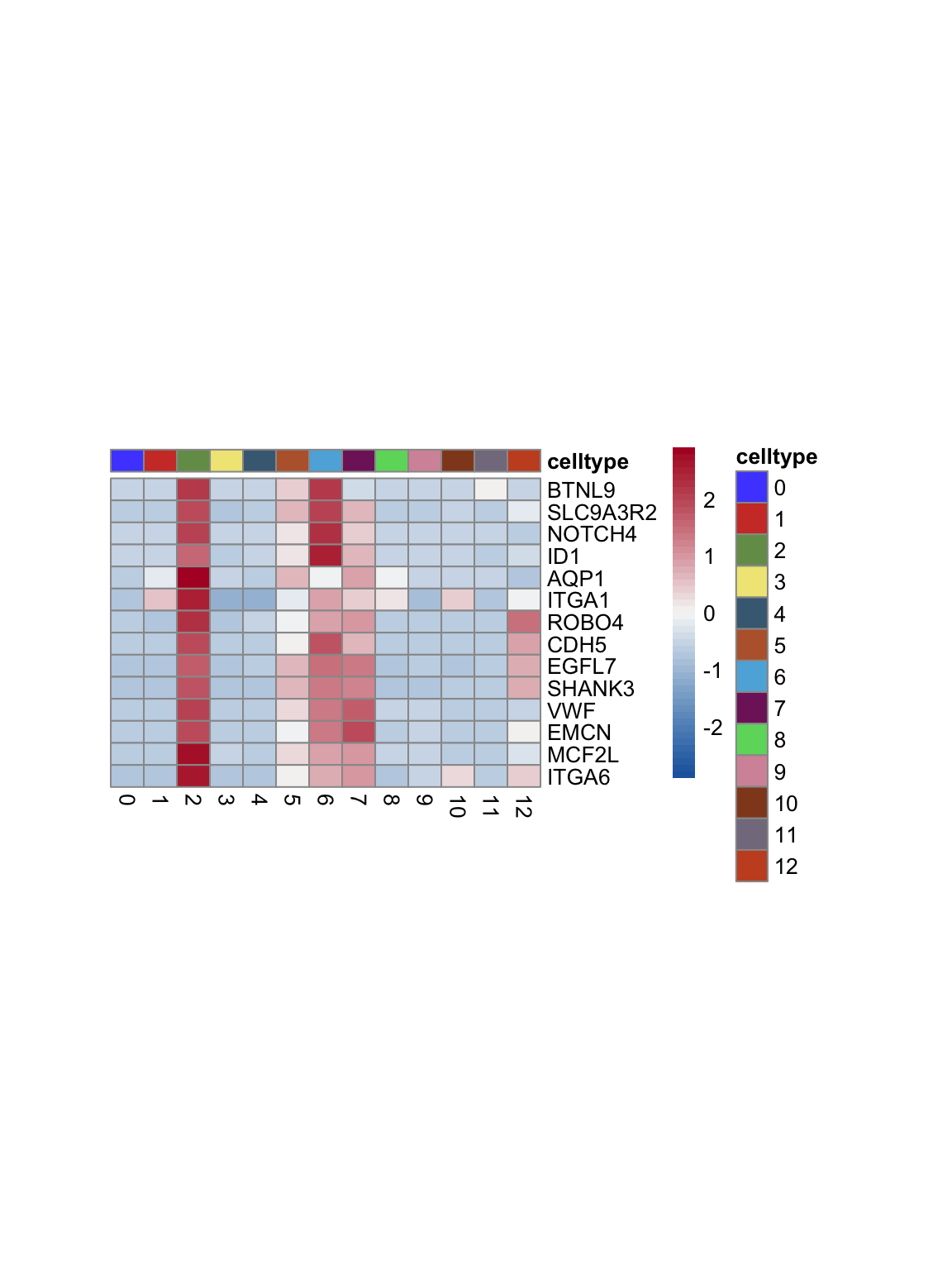
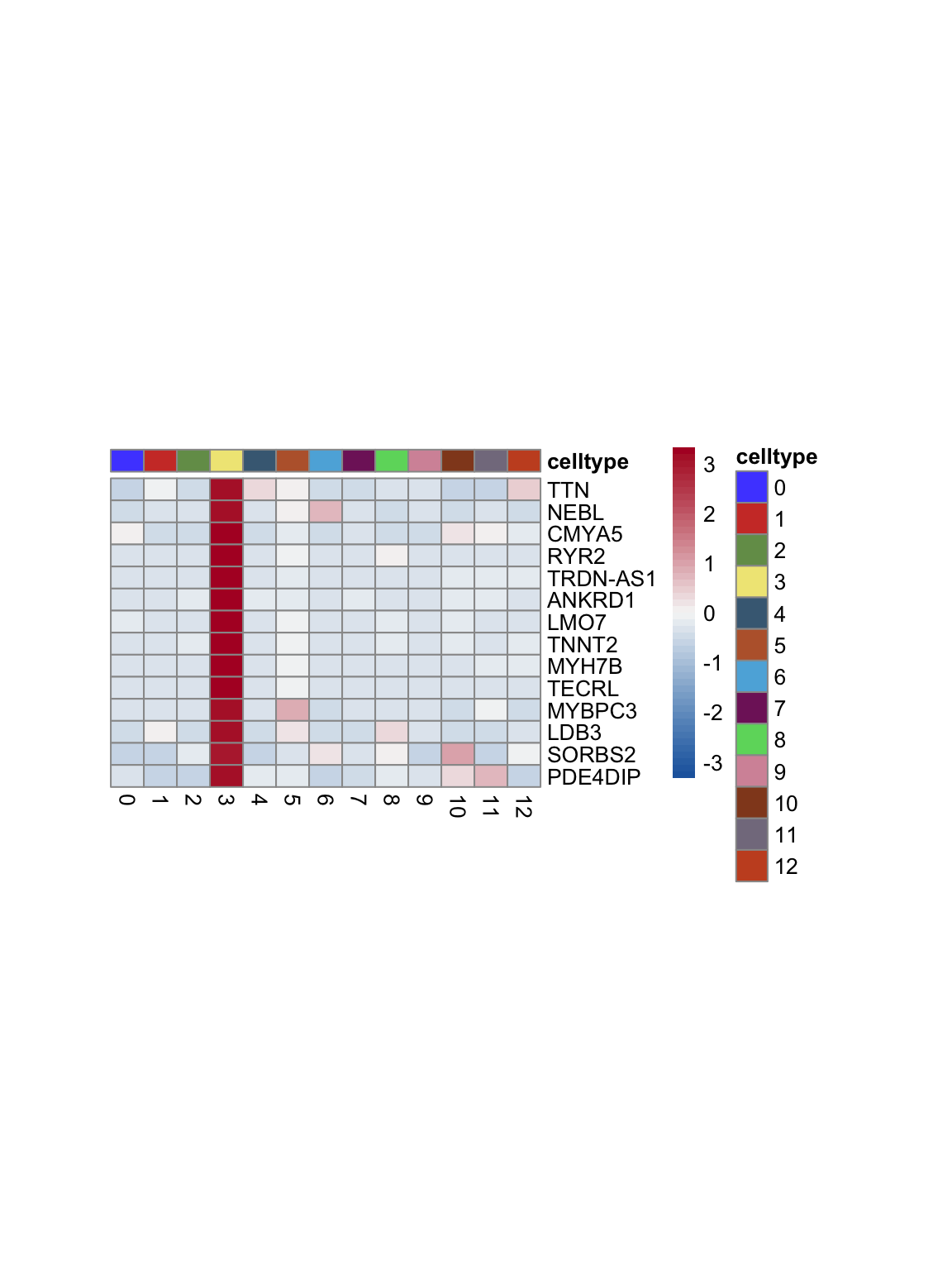
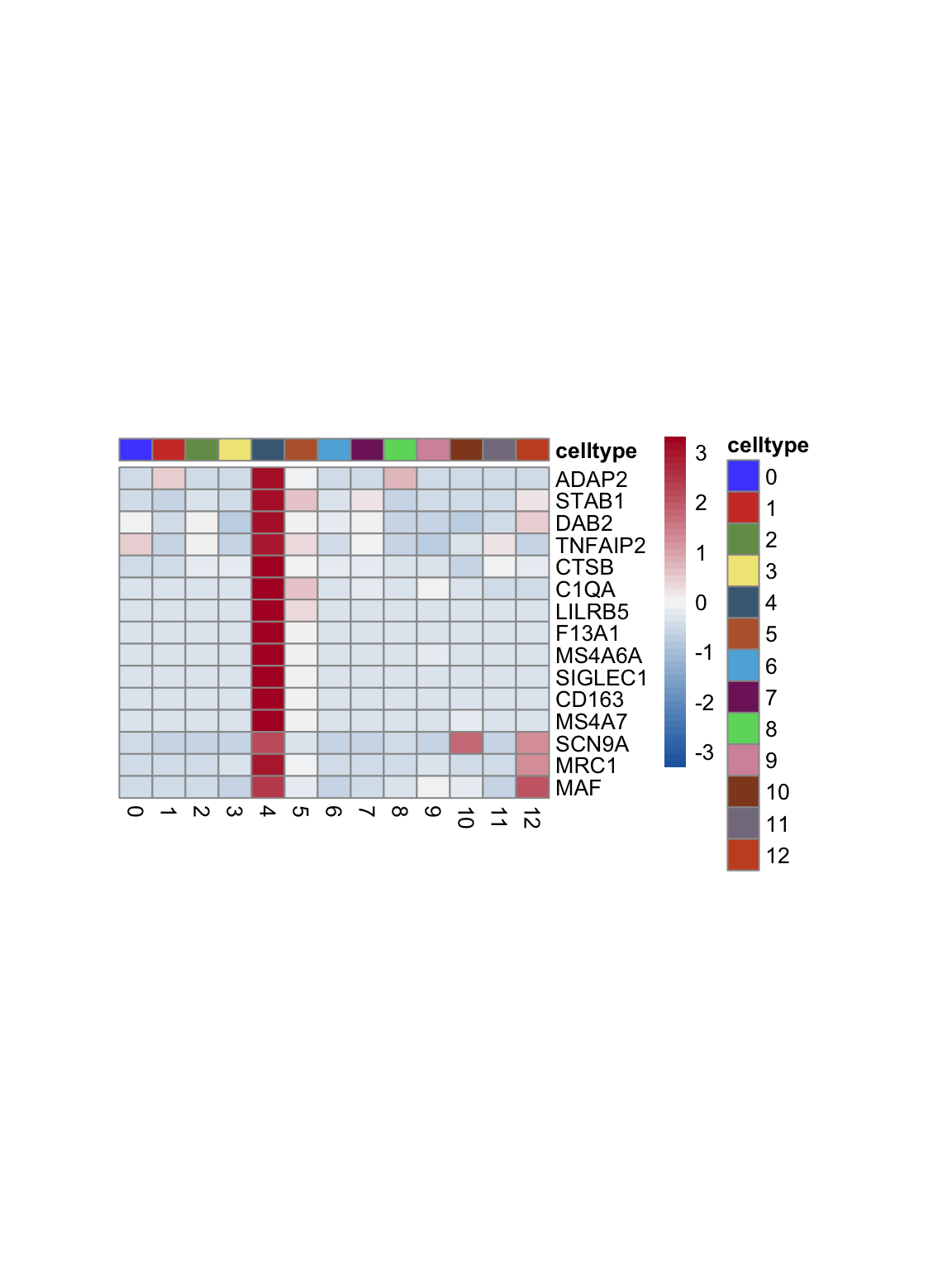
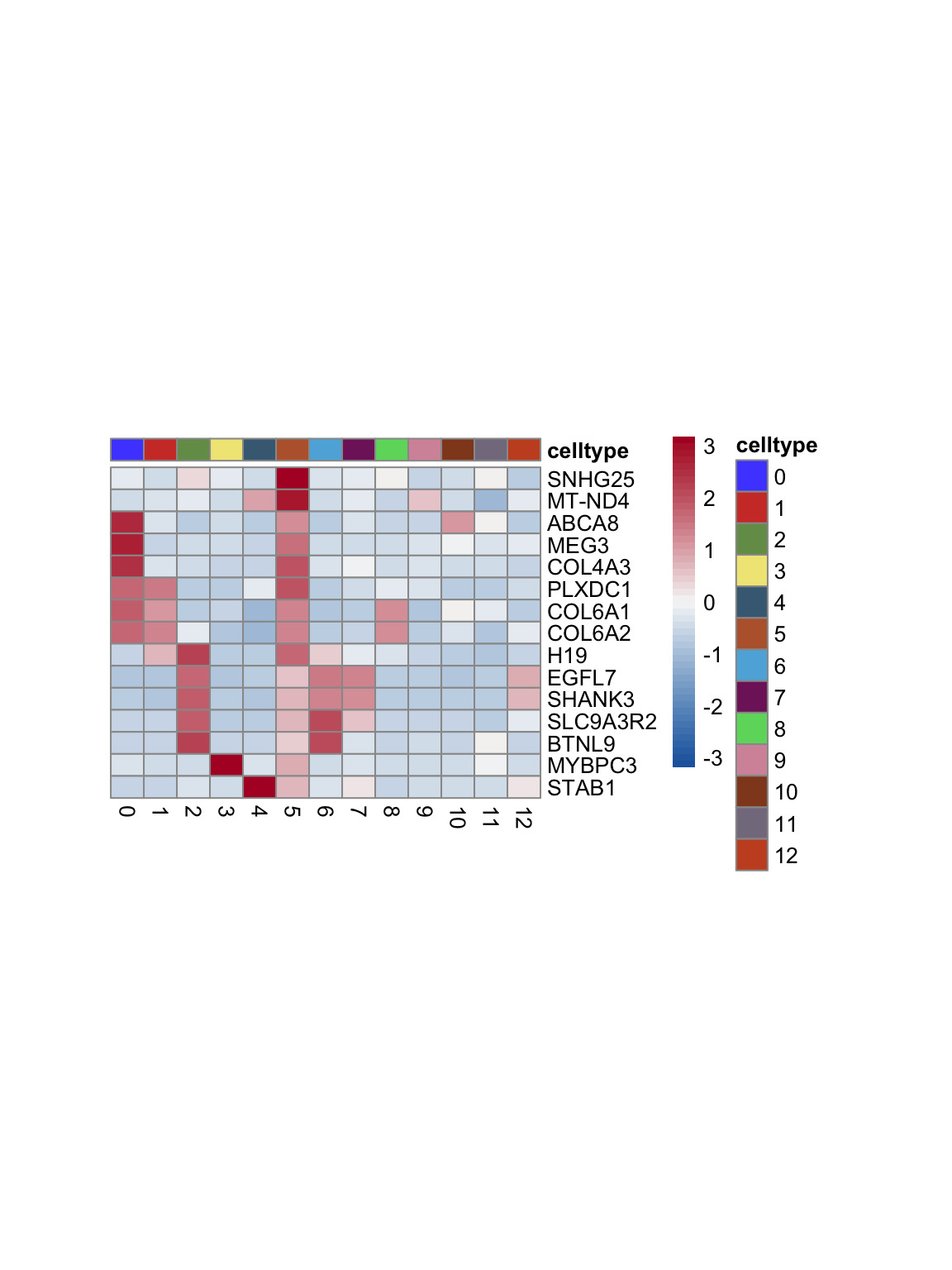
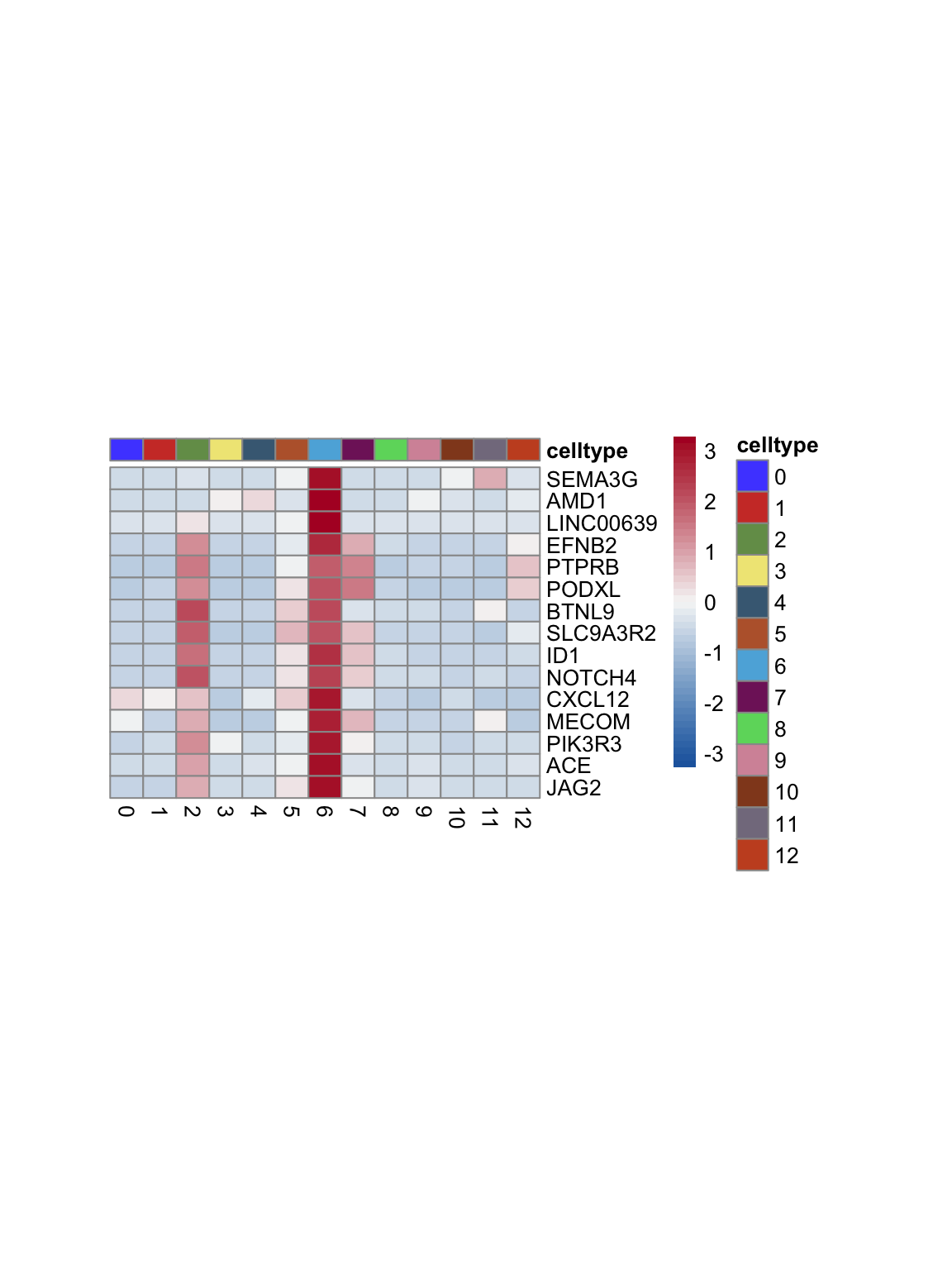
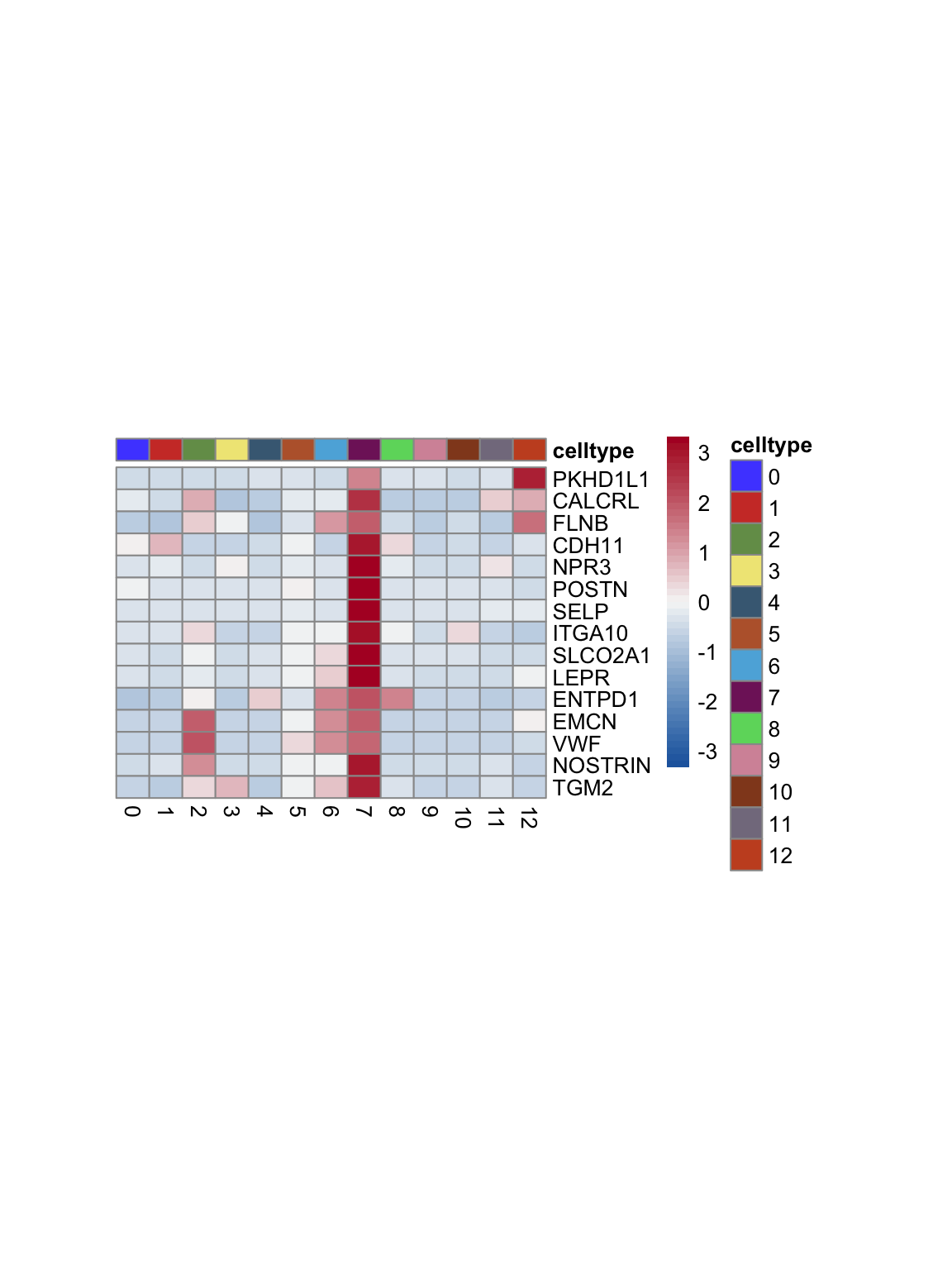
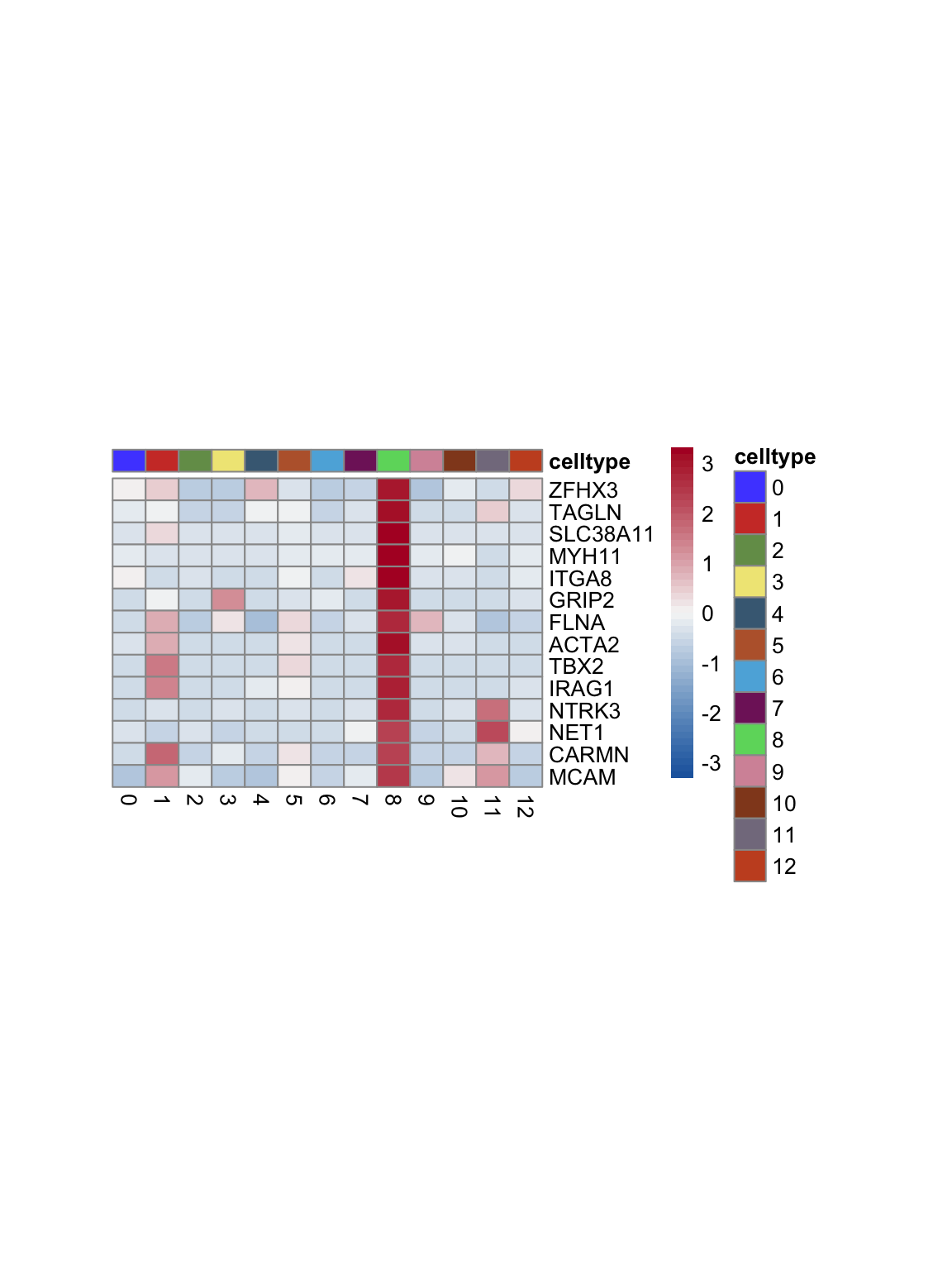
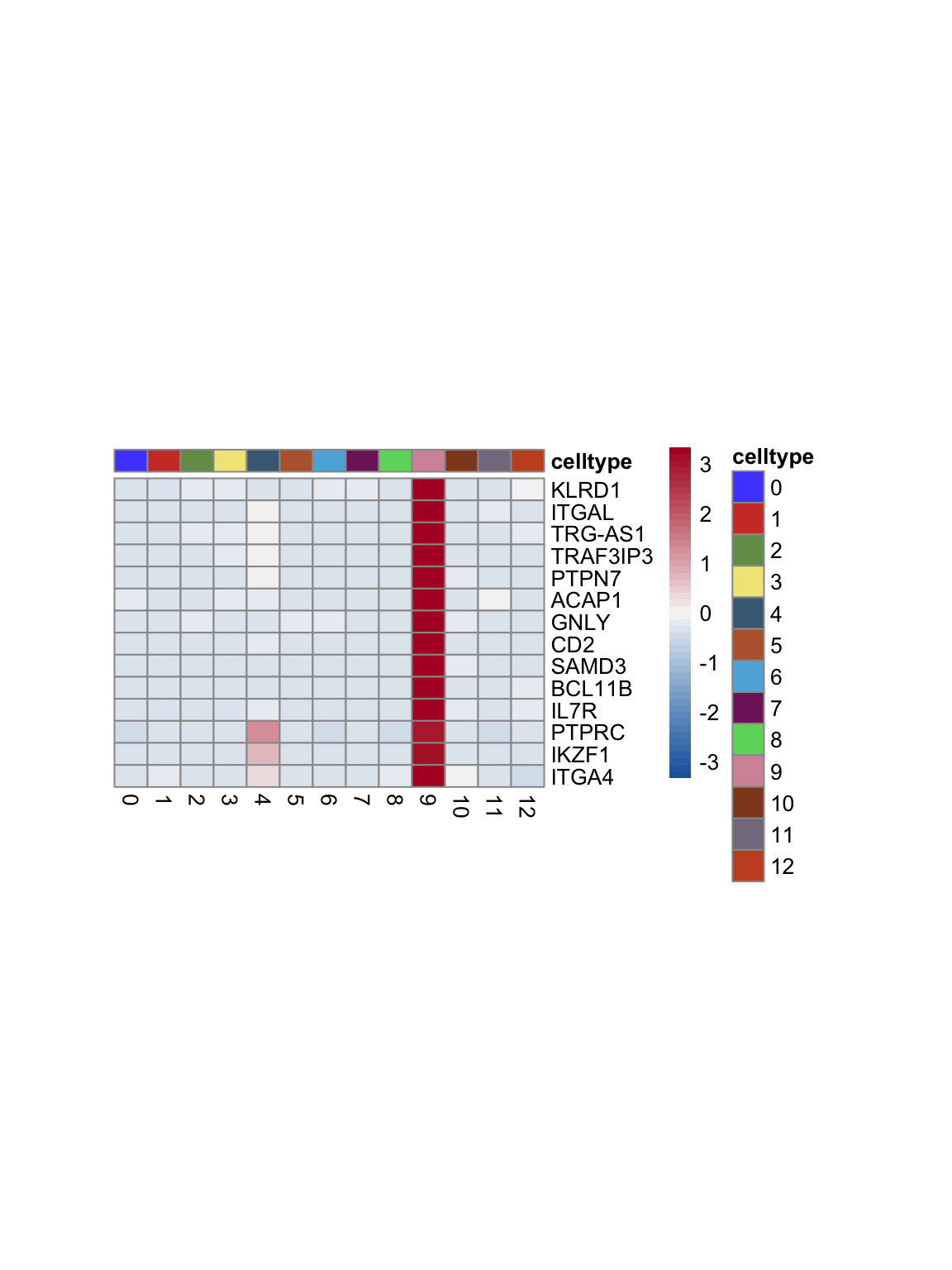
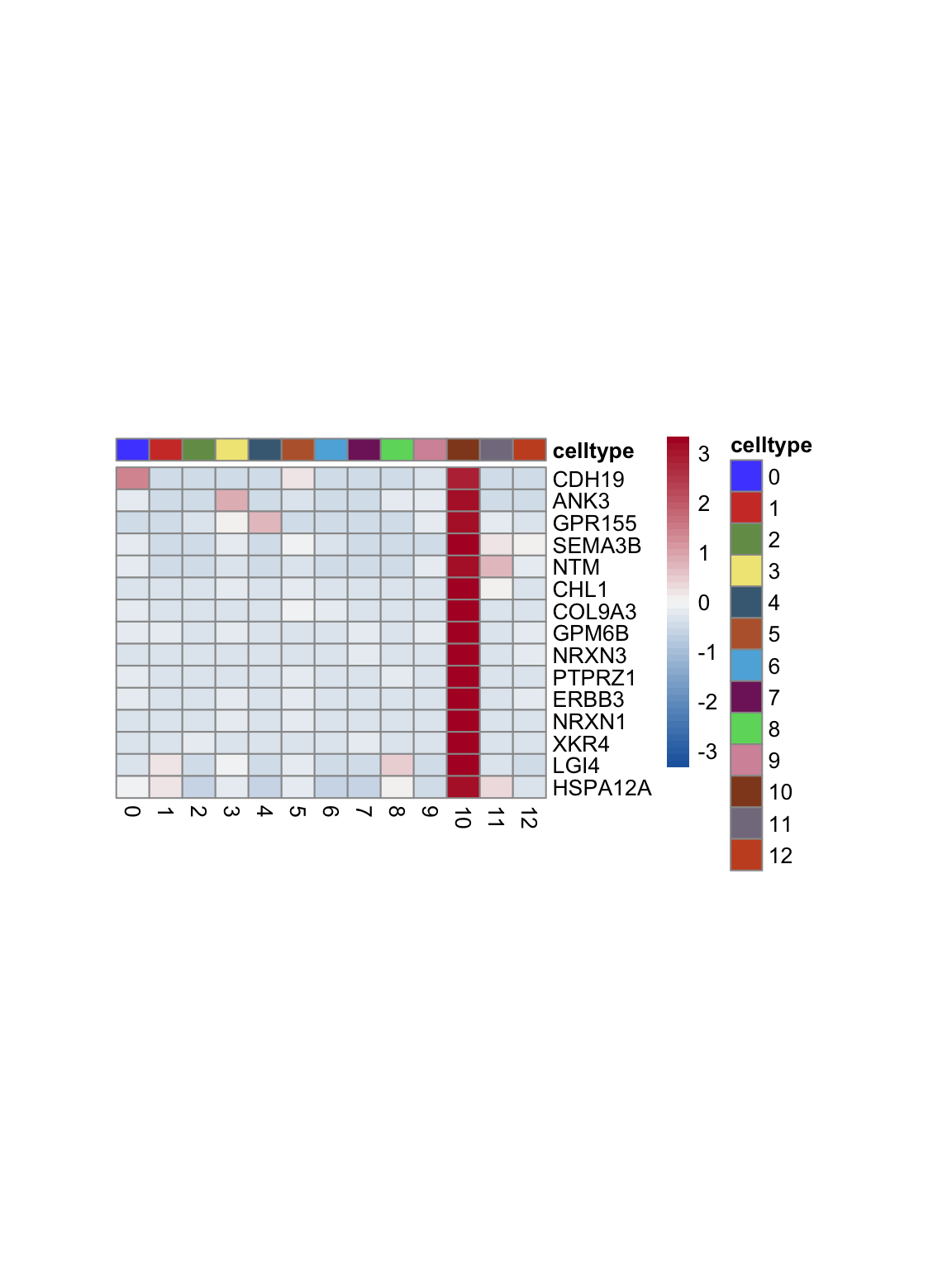
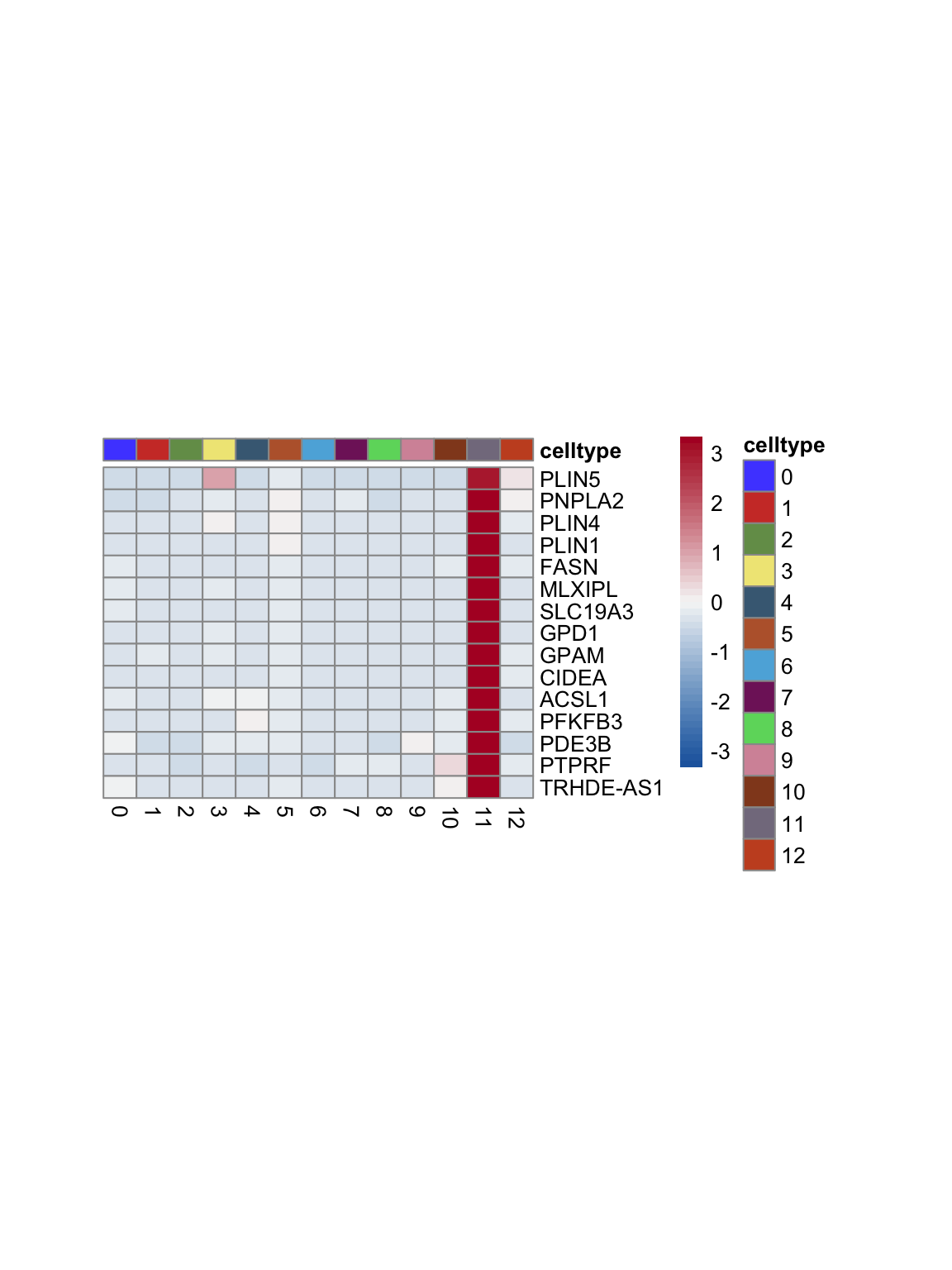
save objects
write.table(seurat_markers_all,
file=paste0(basedir,
"/data/humanHeartsPlusGraz_intPatients_merged",
"_markerGenes.txt"),
row.names = FALSE, col.names = TRUE, quote = FALSE, sep = "\t")session info
sessionInfo()R version 4.2.1 (2022-06-23)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur ... 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] sctransform_0.3.4 viridis_0.6.2
[3] viridisLite_0.4.1 pheatmap_1.0.12
[5] ggpubr_0.4.0 ggsci_2.9
[7] runSeurat3_0.1.0 here_1.0.1
[9] magrittr_2.0.3 sp_1.5-0
[11] SeuratObject_4.1.1 Seurat_4.1.1
[13] forcats_0.5.2 stringr_1.4.1
[15] dplyr_1.0.10 purrr_0.3.4
[17] readr_2.1.2 tidyr_1.2.0
[19] tibble_3.1.8 ggplot2_3.3.6
[21] tidyverse_1.3.2 SingleCellExperiment_1.18.0
[23] SummarizedExperiment_1.26.1 Biobase_2.56.0
[25] GenomicRanges_1.48.0 GenomeInfoDb_1.32.3
[27] IRanges_2.30.1 S4Vectors_0.34.0
[29] BiocGenerics_0.42.0 MatrixGenerics_1.8.1
[31] matrixStats_0.62.0
loaded via a namespace (and not attached):
[1] utf8_1.2.2 reticulate_1.26 tidyselect_1.1.2
[4] htmlwidgets_1.5.4 grid_4.2.1 Rtsne_0.16
[7] munsell_0.5.0 codetools_0.2-18 ica_1.0-3
[10] future_1.28.0 miniUI_0.1.1.1 withr_2.5.0
[13] spatstat.random_2.2-0 colorspace_2.0-3 progressr_0.11.0
[16] highr_0.9 knitr_1.40 rstudioapi_0.14
[19] ROCR_1.0-11 ggsignif_0.6.3 tensor_1.5
[22] listenv_0.8.0 labeling_0.4.2 git2r_0.30.1
[25] GenomeInfoDbData_1.2.8 polyclip_1.10-0 farver_2.1.1
[28] rprojroot_2.0.3 parallelly_1.32.1 vctrs_0.4.1
[31] generics_0.1.3 xfun_0.32 R6_2.5.1
[34] bitops_1.0-7 spatstat.utils_2.3-1 cachem_1.0.6
[37] DelayedArray_0.22.0 assertthat_0.2.1 promises_1.2.0.1
[40] scales_1.2.1 googlesheets4_1.0.1 rgeos_0.5-9
[43] gtable_0.3.1 globals_0.16.1 goftest_1.2-3
[46] workflowr_1.7.0 rlang_1.0.5 splines_4.2.1
[49] rstatix_0.7.0 lazyeval_0.2.2 gargle_1.2.0
[52] spatstat.geom_2.4-0 broom_1.0.1 yaml_2.3.5
[55] reshape2_1.4.4 abind_1.4-5 modelr_0.1.9
[58] backports_1.4.1 httpuv_1.6.5 tools_4.2.1
[61] ellipsis_0.3.2 spatstat.core_2.4-4 jquerylib_0.1.4
[64] RColorBrewer_1.1-3 ggridges_0.5.3 Rcpp_1.0.9
[67] plyr_1.8.7 zlibbioc_1.42.0 RCurl_1.98-1.8
[70] rpart_4.1.16 deldir_1.0-6 pbapply_1.5-0
[73] cowplot_1.1.1 zoo_1.8-10 haven_2.5.1
[76] ggrepel_0.9.1 cluster_2.1.4 fs_1.5.2
[79] data.table_1.14.2 scattermore_0.8 lmtest_0.9-40
[82] reprex_2.0.2 RANN_2.6.1 googledrive_2.0.0
[85] whisker_0.4 fitdistrplus_1.1-8 hms_1.1.2
[88] patchwork_1.1.2 mime_0.12 evaluate_0.16
[91] xtable_1.8-4 readxl_1.4.1 gridExtra_2.3
[94] compiler_4.2.1 KernSmooth_2.23-20 crayon_1.5.1
[97] htmltools_0.5.3 mgcv_1.8-40 later_1.3.0
[100] tzdb_0.3.0 lubridate_1.8.0 DBI_1.1.3
[103] dbplyr_2.2.1 MASS_7.3-58.1 Matrix_1.4-1
[106] car_3.1-0 cli_3.3.0 parallel_4.2.1
[109] igraph_1.3.4 pkgconfig_2.0.3 plotly_4.10.0
[112] spatstat.sparse_2.1-1 xml2_1.3.3 bslib_0.4.0
[115] XVector_0.36.0 rvest_1.0.3 digest_0.6.29
[118] RcppAnnoy_0.0.19 spatstat.data_2.2-0 rmarkdown_2.16
[121] cellranger_1.1.0 leiden_0.4.2 uwot_0.1.14
[124] shiny_1.7.2 lifecycle_1.0.1 nlme_3.1-159
[127] jsonlite_1.8.0 carData_3.0-5 limma_3.52.2
[130] fansi_1.0.3 pillar_1.8.1 lattice_0.20-45
[133] fastmap_1.1.0 httr_1.4.4 survival_3.4-0
[136] glue_1.6.2 png_0.1-7 stringi_1.7.8
[139] sass_0.4.2 irlba_2.3.5 future.apply_1.9.0 date()[1] "Mon Sep 12 12:13:36 2022"
sessionInfo()R version 4.2.1 (2022-06-23)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur ... 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] sctransform_0.3.4 viridis_0.6.2
[3] viridisLite_0.4.1 pheatmap_1.0.12
[5] ggpubr_0.4.0 ggsci_2.9
[7] runSeurat3_0.1.0 here_1.0.1
[9] magrittr_2.0.3 sp_1.5-0
[11] SeuratObject_4.1.1 Seurat_4.1.1
[13] forcats_0.5.2 stringr_1.4.1
[15] dplyr_1.0.10 purrr_0.3.4
[17] readr_2.1.2 tidyr_1.2.0
[19] tibble_3.1.8 ggplot2_3.3.6
[21] tidyverse_1.3.2 SingleCellExperiment_1.18.0
[23] SummarizedExperiment_1.26.1 Biobase_2.56.0
[25] GenomicRanges_1.48.0 GenomeInfoDb_1.32.3
[27] IRanges_2.30.1 S4Vectors_0.34.0
[29] BiocGenerics_0.42.0 MatrixGenerics_1.8.1
[31] matrixStats_0.62.0
loaded via a namespace (and not attached):
[1] utf8_1.2.2 reticulate_1.26 tidyselect_1.1.2
[4] htmlwidgets_1.5.4 grid_4.2.1 Rtsne_0.16
[7] munsell_0.5.0 codetools_0.2-18 ica_1.0-3
[10] future_1.28.0 miniUI_0.1.1.1 withr_2.5.0
[13] spatstat.random_2.2-0 colorspace_2.0-3 progressr_0.11.0
[16] highr_0.9 knitr_1.40 rstudioapi_0.14
[19] ROCR_1.0-11 ggsignif_0.6.3 tensor_1.5
[22] listenv_0.8.0 labeling_0.4.2 git2r_0.30.1
[25] GenomeInfoDbData_1.2.8 polyclip_1.10-0 farver_2.1.1
[28] rprojroot_2.0.3 parallelly_1.32.1 vctrs_0.4.1
[31] generics_0.1.3 xfun_0.32 R6_2.5.1
[34] bitops_1.0-7 spatstat.utils_2.3-1 cachem_1.0.6
[37] DelayedArray_0.22.0 assertthat_0.2.1 promises_1.2.0.1
[40] scales_1.2.1 googlesheets4_1.0.1 rgeos_0.5-9
[43] gtable_0.3.1 globals_0.16.1 goftest_1.2-3
[46] workflowr_1.7.0 rlang_1.0.5 splines_4.2.1
[49] rstatix_0.7.0 lazyeval_0.2.2 gargle_1.2.0
[52] spatstat.geom_2.4-0 broom_1.0.1 yaml_2.3.5
[55] reshape2_1.4.4 abind_1.4-5 modelr_0.1.9
[58] backports_1.4.1 httpuv_1.6.5 tools_4.2.1
[61] ellipsis_0.3.2 spatstat.core_2.4-4 jquerylib_0.1.4
[64] RColorBrewer_1.1-3 ggridges_0.5.3 Rcpp_1.0.9
[67] plyr_1.8.7 zlibbioc_1.42.0 RCurl_1.98-1.8
[70] rpart_4.1.16 deldir_1.0-6 pbapply_1.5-0
[73] cowplot_1.1.1 zoo_1.8-10 haven_2.5.1
[76] ggrepel_0.9.1 cluster_2.1.4 fs_1.5.2
[79] data.table_1.14.2 scattermore_0.8 lmtest_0.9-40
[82] reprex_2.0.2 RANN_2.6.1 googledrive_2.0.0
[85] whisker_0.4 fitdistrplus_1.1-8 hms_1.1.2
[88] patchwork_1.1.2 mime_0.12 evaluate_0.16
[91] xtable_1.8-4 readxl_1.4.1 gridExtra_2.3
[94] compiler_4.2.1 KernSmooth_2.23-20 crayon_1.5.1
[97] htmltools_0.5.3 mgcv_1.8-40 later_1.3.0
[100] tzdb_0.3.0 lubridate_1.8.0 DBI_1.1.3
[103] dbplyr_2.2.1 MASS_7.3-58.1 Matrix_1.4-1
[106] car_3.1-0 cli_3.3.0 parallel_4.2.1
[109] igraph_1.3.4 pkgconfig_2.0.3 plotly_4.10.0
[112] spatstat.sparse_2.1-1 xml2_1.3.3 bslib_0.4.0
[115] XVector_0.36.0 rvest_1.0.3 digest_0.6.29
[118] RcppAnnoy_0.0.19 spatstat.data_2.2-0 rmarkdown_2.16
[121] cellranger_1.1.0 leiden_0.4.2 uwot_0.1.14
[124] shiny_1.7.2 lifecycle_1.0.1 nlme_3.1-159
[127] jsonlite_1.8.0 carData_3.0-5 limma_3.52.2
[130] fansi_1.0.3 pillar_1.8.1 lattice_0.20-45
[133] fastmap_1.1.0 httr_1.4.4 survival_3.4-0
[136] glue_1.6.2 png_0.1-7 stringi_1.7.8
[139] sass_0.4.2 irlba_2.3.5 future.apply_1.9.0