library(tidyverse)
library(SpatialExperiment)
library(here)
library(ggspavis)
library(scater)
library(ggsci)
library(scran)
library(pheatmap)
library(SpaTalk)
library(Matrix)
library(Seurat)
library(igraph)
library(biomaRt)Run spatalk decomposition - s4 batch2
Load packages
Sample 4
Load reference and spe object
basedir <- here()
refSeurat <- readRDS(paste0(basedir, "/data/refSeurat_allCelltypesIntImm.rds"))
speAll <- readRDS(paste0(basedir, "/data/speAllLNB2.rds"))
genes <- data.frame(geneLong=rownames(refSeurat)) %>%
mutate(EnsID=gsub("\\..*", "", geneLong)) %>%
mutate(symbol=substr(geneLong, start=17, stop=nchar(geneLong)))set color palettes
colLEC <- c("#79AF97FF","#B24745FF","#DF8F44FF")
names(colLEC) <- c("SCScLEC", "SCSfLEC", "MedSinusLEC")
colBEC <- c("#48557e", "#e3a616","#9a3038")
names(colBEC) <- c("venBEC", "capBEC", "artBEC")
colFRC <- c("#800000FF", "#FFA319FF","#8A9045FF", "#155F83FF",
"#C16622FF", "#6692a3", "#3b7f60")
names(colFRC) <- c("medRCIFRC", "TRC", "ACTA2+PRC", "VSMC", "PI16+RC", "PRC1",
"BRC")
colImm <- c("#0b6647", "#54907e", "#94c78a", "#6f9568",
"#8f2810", "#d0ac21","#9e9f0b", "#486584",
"#4b5397", "#8873d3", "#6e3e7a")
names(colImm) <- c("naiveB", "GCB", "MBC", "plasmaCell",
"CD4T", "CD8Tcm", "CTL/NKcell", "ILC3", "pDC", "Mph/DC-1",
"Mph/DC-2")
colAll <- c(colFRC, colBEC, colLEC, colImm)
colPal <- c("#F0027F", "#377EB8", "#4DAF4A", "#984EA3", "#FFD700",
"#FF7F00", "#1A1A1A", "#666666", pal_futurama()(10))format sc reference data
## downsample
dim(refSeurat)[1] 39642 122538table(refSeurat$label)
ACTA2+PRC artBEC BRC capBEC CD4T CD8Tcm
1560 1048 390 1091 39380 4006
CTL/NKcell GCB ILC3 MBC medRCIFRC MedSinusLEC
10339 869 1819 17174 10129 42
Mph/DC-1 Mph/DC-2 naiveB pDC PI16+RC plasmaCell
2952 980 14118 6750 986 326
PRC1 SCScLEC SCSfLEC TRC venBEC VSMC
760 332 129 3773 2861 724 #refSeurat$pat_plus_label <- paste0(refSeurat$patient, "_", refSeurat$label)
#table(refSeurat$pat_plus_label)
#Idents(refSeurat) <- refSeurat$pat_plus_label
Idents(refSeurat) <- refSeurat$label
refSeurat <- subset(x = refSeurat, downsample = 500)
dim(refSeurat)[1] 39642 10719#table(refSeurat$pat_plus_label)
table(refSeurat$label)
ACTA2+PRC artBEC BRC capBEC CD4T CD8Tcm
500 500 390 500 500 500
CTL/NKcell GCB ILC3 MBC medRCIFRC MedSinusLEC
500 500 500 500 500 42
Mph/DC-1 Mph/DC-2 naiveB pDC PI16+RC plasmaCell
500 500 500 500 500 326
PRC1 SCScLEC SCSfLEC TRC venBEC VSMC
500 332 129 500 500 500 ## counts
sc_data <- refSeurat@assays$RNA@counts
rownames(sc_data) <- substr(rownames(sc_data), start=17,
stop=nchar(rownames(sc_data)))
## celltypes
sc_celltype <- refSeurat$label
## remove CC marker
CCmarker <- c(cc.genes.updated.2019$s.genes, cc.genes.updated.2019$g2m.genes)
dim(sc_data)[1] 39642 10719sc_data <- sc_data[-which(rownames(sc_data) %in% CCmarker),]
dim(sc_data)[1] 39545 10719## remove Mito and Ribosomal genes from reference
mito.genes <- grep(pattern = "^MT-", x = rownames(sc_data), value = TRUE)
RPS.genes <- grep(pattern = "^RPS", x = rownames(sc_data), value = TRUE)
RPL.genes <- grep(pattern = "^RPL", x = rownames(sc_data), value = TRUE)
dim(sc_data)[1] 39545 10719sc_data <- sc_data[-which(rownames(sc_data) %in% c(mito.genes, RPS.genes,
RPL.genes)),]
dim(sc_data)[1] 38478 10719vis input data
ref data
Idents(refSeurat) <- refSeurat$label
DimPlot(refSeurat, reduction = "umap", cols = colAll, raster = F, shuffle = T)+
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank(),
panel.grid.minor = element_blank()) +
xlab("UMAP1") +
ylab("UMAP2")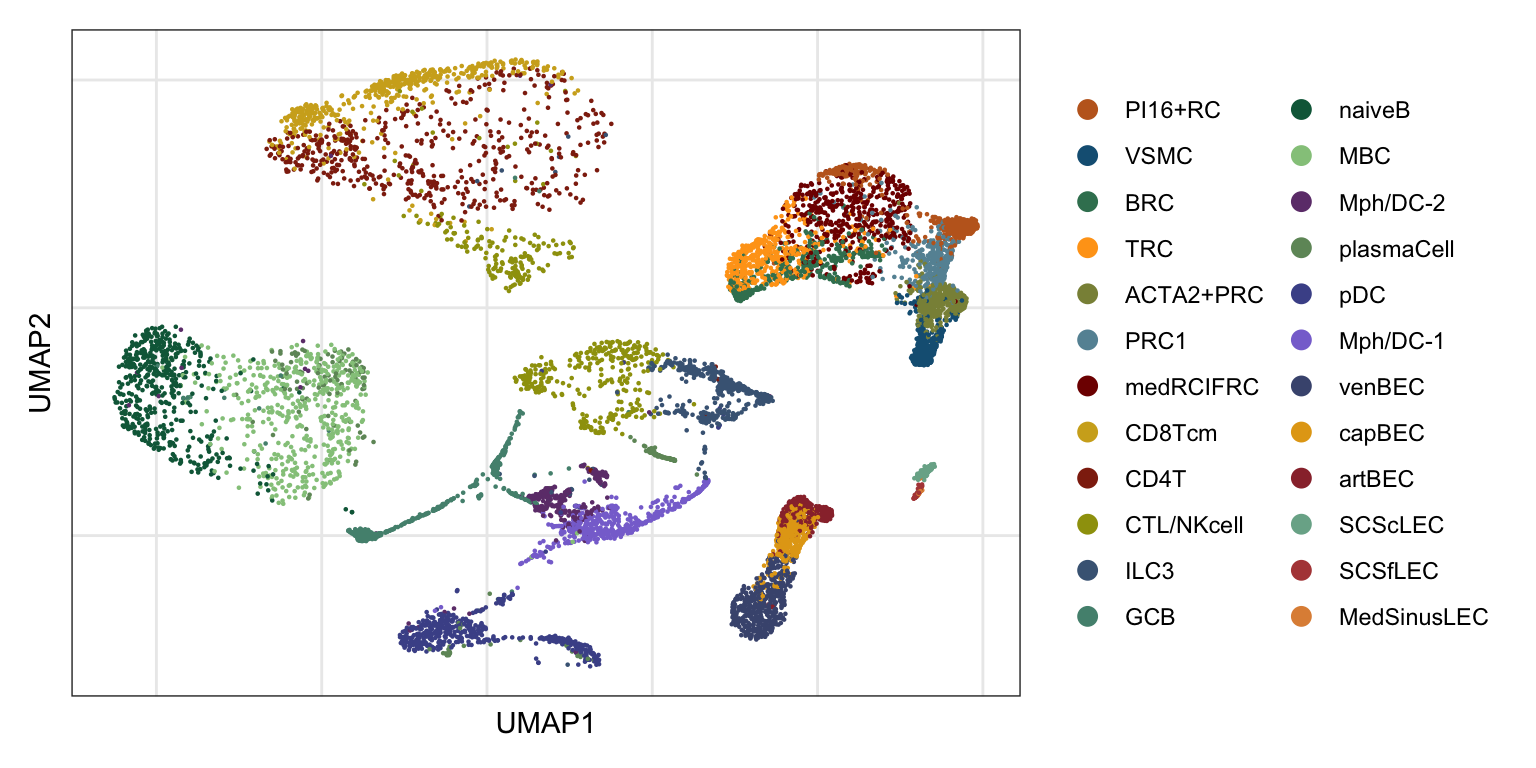
remove(refSeurat)ST data
i <- 4
sid <- names(speAll[i])
spe <- speAll[[i]]
plotSpots(spe, annotate = "label",
palette = colPal, size = 0.8) +
ggtitle(sid)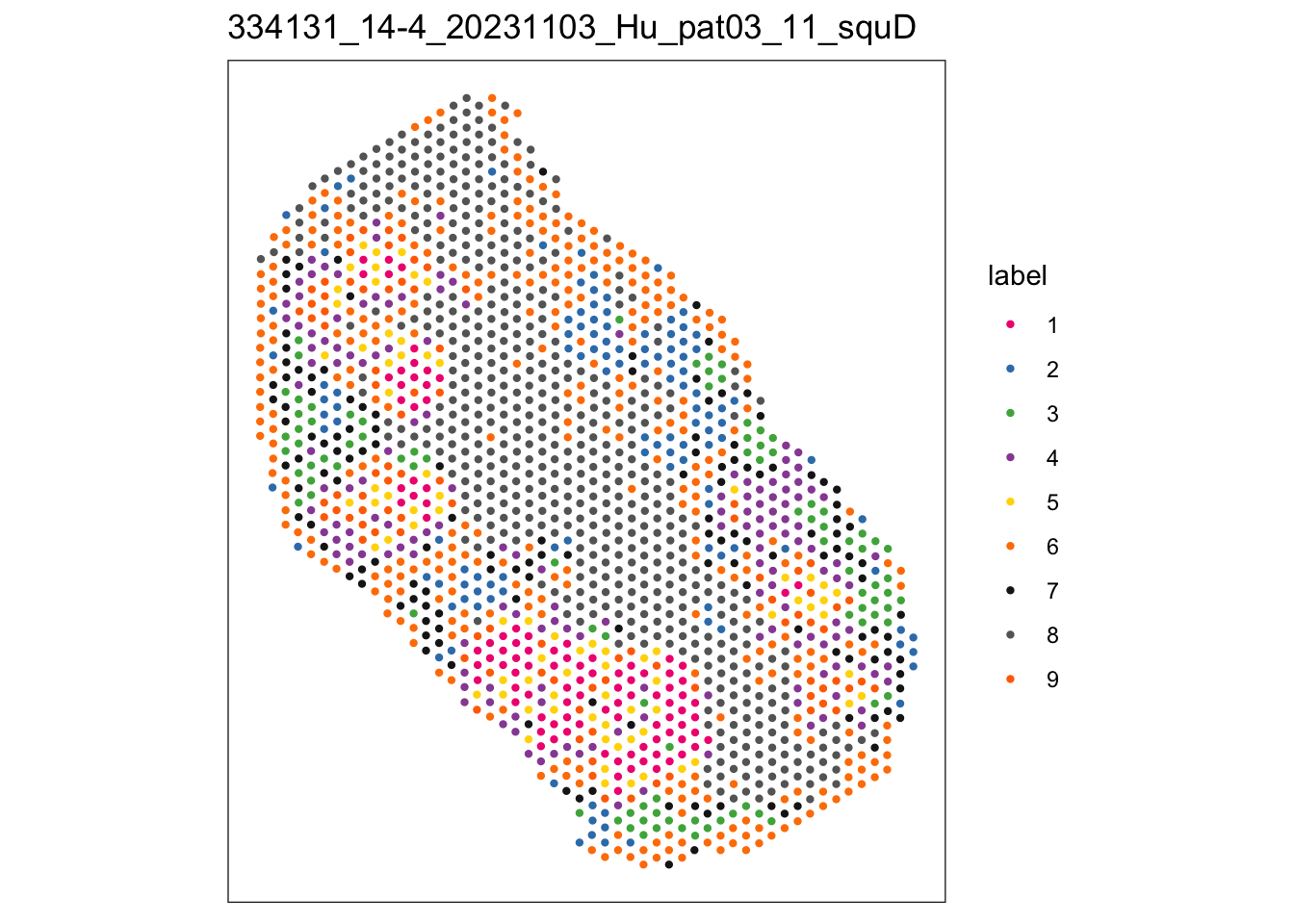
remove(speAll)format spatial data
## counts
st_data <- spe@assays@data$counts
## transform gene names
genesST <- data.frame(EnsID=rownames(st_data)) %>%
left_join(., genes, by="EnsID")
mart <- useDataset("hsapiens_gene_ensembl", useMart("ensembl"))
gene_IDs <- getBM(filters= "ensembl_gene_id", attributes= c("ensembl_gene_id","hgnc_symbol"),
values = genesST$EnsID, mart= mart, useCache = F)
colnames(gene_IDs) <- c("EnsID", "symbolMart")
gene_IDs <- gene_IDs[-which(duplicated(gene_IDs$EnsID)),]
genesST <- genesST %>% left_join(., gene_IDs, by="EnsID") %>%
mutate(symbolFin=ifelse(is.na(symbol),ifelse(symbolMart =="", EnsID, symbolMart),symbol))
rownames(st_data) <- genesST$symbolFin
st_data <- rev_gene(data = st_data,
data_type = "count",
species = "Human",
geneinfo = geneinfo)
## coords
st_meta <- data.frame(spot=colnames(spe),
x=spe@int_colData@listData$spatialCoords[,"pxl_col_in_fullres"],
y=-1*(spe@int_colData@listData$spatialCoords[,"pxl_row_in_fullres"]))
obj <- createSpaTalk(st_data = as.matrix(st_data),
st_meta = st_meta,
species = "Human",
if_st_is_sc = F,
spot_max_cell = 30)save object to run CT on server
celltype decomposition
remove(st_data)
remove(st_meta)
remove(mart)
obj <- dec_celltype(object = obj,
sc_data = sc_data,
sc_celltype = sc_celltype,
if_use_hvg = T,
use_n_cores = 6,
iter_num = 100,
if_use_normalize_data = F)
saveRDS(obj, file=paste0(basedir, "/data/ST/spatalk/spatalkDecompB2_s4.rds"))Session info
sessionInfo()R version 4.3.0 (2023-04-21)
Platform: x86_64-apple-darwin20 (64-bit)
Running under: macOS Ventura 13.4.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Europe/Berlin
tzcode source: internal
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] biomaRt_2.56.1 igraph_2.0.2
[3] Seurat_5.0.2 SeuratObject_5.0.1
[5] sp_2.1-3 Matrix_1.6-5
[7] SpaTalk_1.0 doParallel_1.0.17
[9] iterators_1.0.14 foreach_1.5.2
[11] ggalluvial_0.12.5 pheatmap_1.0.12
[13] scran_1.28.2 ggsci_3.0.1
[15] scater_1.28.0 scuttle_1.10.3
[17] ggspavis_1.6.0 here_1.0.1
[19] SpatialExperiment_1.10.0 SingleCellExperiment_1.22.0
[21] SummarizedExperiment_1.30.2 Biobase_2.60.0
[23] GenomicRanges_1.52.1 GenomeInfoDb_1.36.4
[25] IRanges_2.36.0 S4Vectors_0.40.1
[27] BiocGenerics_0.48.0 MatrixGenerics_1.12.3
[29] matrixStats_1.2.0 lubridate_1.9.3
[31] forcats_1.0.0 stringr_1.5.1
[33] dplyr_1.1.4 purrr_1.0.2
[35] readr_2.1.5 tidyr_1.3.1
[37] tibble_3.2.1 ggplot2_3.5.0
[39] tidyverse_2.0.0
loaded via a namespace (and not attached):
[1] spatstat.sparse_3.0-3 bitops_1.0-7
[3] httr_1.4.7 RColorBrewer_1.1-3
[5] backports_1.4.1 tools_4.3.0
[7] sctransform_0.4.1 utf8_1.2.4
[9] R6_2.5.1 HDF5Array_1.28.1
[11] lazyeval_0.2.2 uwot_0.1.16
[13] rhdf5filters_1.12.1 withr_3.0.0
[15] prettyunits_1.2.0 gridExtra_2.3
[17] progressr_0.14.0 cli_3.6.2
[19] spatstat.explore_3.2-6 fastDummies_1.7.3
[21] scatterpie_0.2.1 labeling_0.4.3
[23] spatstat.data_3.0-4 ggridges_0.5.6
[25] pbapply_1.7-2 R.utils_2.12.3
[27] parallelly_1.37.1 limma_3.56.2
[29] RSQLite_2.3.5 rstudioapi_0.15.0
[31] generics_0.1.3 ica_1.0-3
[33] spatstat.random_3.2-3 car_3.1-2
[35] ggbeeswarm_0.7.2 fansi_1.0.6
[37] abind_1.4-5 R.methodsS3_1.8.2
[39] lifecycle_1.0.4 yaml_2.3.8
[41] edgeR_3.42.4 carData_3.0-5
[43] BiocFileCache_2.8.0 rhdf5_2.44.0
[45] Rtsne_0.17 blob_1.2.4
[47] grid_4.3.0 promises_1.2.1
[49] dqrng_0.3.2 crayon_1.5.2
[51] miniUI_0.1.1.1 lattice_0.22-5
[53] beachmat_2.16.0 cowplot_1.1.3
[55] KEGGREST_1.40.1 magick_2.8.3
[57] pillar_1.9.0 knitr_1.45
[59] metapod_1.8.0 rjson_0.2.21
[61] future.apply_1.11.1 codetools_0.2-19
[63] leiden_0.4.3.1 glue_1.7.0
[65] ggfun_0.1.4 data.table_1.15.2
[67] vctrs_0.6.5 png_0.1-8
[69] spam_2.10-0 gtable_0.3.4
[71] cachem_1.0.8 xfun_0.42
[73] S4Arrays_1.0.6 mime_0.12
[75] DropletUtils_1.20.0 NNLM_0.4.4
[77] ggside_0.3.1 survival_3.5-8
[79] statmod_1.5.0 bluster_1.10.0
[81] ellipsis_0.3.2 fitdistrplus_1.1-11
[83] ROCR_1.0-11 nlme_3.1-164
[85] bit64_4.0.5 filelock_1.0.3
[87] progress_1.2.3 RcppAnnoy_0.0.22
[89] rprojroot_2.0.4 irlba_2.3.5.1
[91] vipor_0.4.7 KernSmooth_2.23-22
[93] DBI_1.2.2 colorspace_2.1-0
[95] tidyselect_1.2.0 curl_5.2.1
[97] bit_4.0.5 compiler_4.3.0
[99] BiocNeighbors_1.18.0 xml2_1.3.6
[101] DelayedArray_0.26.7 plotly_4.10.4
[103] scales_1.3.0 lmtest_0.9-40
[105] rappdirs_0.3.3 digest_0.6.34
[107] goftest_1.2-3 spatstat.utils_3.0-4
[109] rmarkdown_2.26 XVector_0.40.0
[111] htmltools_0.5.7 pkgconfig_2.0.3
[113] sparseMatrixStats_1.12.2 dbplyr_2.4.0
[115] fastmap_1.1.1 rlang_1.1.3
[117] htmlwidgets_1.6.4 shiny_1.8.0
[119] DelayedMatrixStats_1.22.6 farver_2.1.1
[121] zoo_1.8-12 jsonlite_1.8.8
[123] BiocParallel_1.34.2 R.oo_1.26.0
[125] BiocSingular_1.16.0 RCurl_1.98-1.14
[127] magrittr_2.0.3 GenomeInfoDbData_1.2.10
[129] dotCall64_1.1-1 patchwork_1.2.0
[131] Rhdf5lib_1.22.1 munsell_0.5.0
[133] Rcpp_1.0.12 viridis_0.6.5
[135] reticulate_1.35.0 stringi_1.8.3
[137] zlibbioc_1.46.0 MASS_7.3-60.0.1
[139] plyr_1.8.9 listenv_0.9.1
[141] ggrepel_0.9.5 deldir_2.0-4
[143] Biostrings_2.68.1 splines_4.3.0
[145] tensor_1.5 hms_1.1.3
[147] locfit_1.5-9.9 ggpubr_0.6.0
[149] spatstat.geom_3.2-9 ggsignif_0.6.4
[151] RcppHNSW_0.6.0 reshape2_1.4.4
[153] ScaledMatrix_1.8.1 XML_3.99-0.16.1
[155] evaluate_0.23 tzdb_0.4.0
[157] tweenr_2.0.3 httpuv_1.6.14
[159] RANN_2.6.1 polyclip_1.10-6
[161] future_1.33.1 scattermore_1.2
[163] ggforce_0.4.2 ggExtra_0.10.1
[165] rsvd_1.0.5 broom_1.0.5
[167] xtable_1.8-4 RSpectra_0.16-1
[169] rstatix_0.7.2 later_1.3.2
[171] viridisLite_0.4.2 memoise_2.0.1
[173] AnnotationDbi_1.62.2 beeswarm_0.4.0
[175] cluster_2.1.6 timechange_0.3.0
[177] globals_0.16.2 date()[1] "Wed Mar 13 22:10:36 2024"